Citation: Anna Kahler, Heinrich Sticht. A modeling strategy for G-protein coupled receptors[J]. AIMS Biophysics, 2016, 3(2): 211-231. doi: 10.3934/biophy.2016.2.211

| [1] |
Takata K, Matsuzaki T, Tajika Y (2004) Aquaporins: water channel proteins of the cell membrane. Prog Histochem Cytochem 39: 1–83. doi: 10.1016/j.proghi.2004.03.001

|
| [2] |
Nagel G, Szellas T, Huhn W, et al. (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A 100: 13940–13945. doi: 10.1073/pnas.1936192100
|
| [3] |
Lai EC (2004) Notch signaling: control of cell communication and cell fate. Development 131: 965–973. doi: 10.1242/dev.01074
|
| [4] |
Jacoby E, Bouhelal R, Gerspacher M, et al. (2006) The 7 TM G-Protein-Coupled Receptor Target Family. ChemMedChem 1: 760–782. doi: 10.1002/cmdc.200600134
|
| [5] |
Gentry PR, Sexton PM, Christopoulos A (2015) Novel Allosteric Modulators of G Protein-coupled Receptors. J Biol Chem 290: 19478–19488. doi: 10.1074/jbc.R115.662759
|
| [6] |
Strotmann R, Schröck K, Böselt I, et al. (2011) Evolution of GPCR: Change and continuity. Mol Cell Endocrinol 331: 170–178. doi: 10.1016/j.mce.2010.07.012
|
| [7] |
Ferré S (2015) The GPCR heterotetramer: challenging classical pharmacology. Trends Pharmacol Sci 36: 145–152. doi: 10.1016/j.tips.2015.01.002
|
| [8] | Joost P, Methner A (2002) Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biology 3: research0063.1–research0063.16. |
| [9] |
Kledal TN, Rosenkilde MM, Schwartz TW (1998) Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Letters 441: 209–214. doi: 10.1016/S0014-5793(98)01551-8
|
| [10] |
Tan Q, Zhu Y, Li J, et al. (2013) Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 341: 1387–1390. doi: 10.1126/science.1241475
|
| [11] |
Sali A, Blundell TL (1993) Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol 234: 779–815. doi: 10.1006/jmbi.1993.1626
|
| [12] |
Wu B, Chien EYT, Mol CD, et al. (2010) Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 330: 1066–1071. doi: 10.1126/science.1194396
|
| [13] | Park SH, Das BB, Casagrande F, et al. (2012) Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491: 779–783. |
| [14] |
Burg JS, Ingram JR, Venkatakrishnan AJ, et al. (2015) Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347: 1113–1117. doi: 10.1126/science.aaa5026
|
| [15] | Bateman A, Birney E, Durbin R, et al. (2000) The Pfam Protein Families Database. |
| [16] | Eddy SR (2009) A new Generation of Homology Search Tools based on Probabilistic Inference. Genome Inform 23: 205–211. |
| [17] |
Johnson LS, Eddy S, Portugaly E (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11: 431. doi: 10.1186/1471-2105-11-431
|
| [18] |
Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195. doi: 10.1371/journal.pcbi.1002195
|
| [19] | Hooft RWW, Vriend G, Sander C, et al. (1996) Errors in protein structures. Nature 381: 272. |
| [20] |
Dolinsky TJ, Nielsen JE, McCammon JA, et al. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32: W665–W667. doi: 10.1093/nar/gkh381
|
| [21] |
Berendsen HJC, Postma JPM, van Gunsteren WF, et al. (1981) Interaction models for water in relation to protein hydration. Intermolecular Forces 14: 331–342. doi: 10.1007/978-94-015-7658-1_21
|
| [22] |
Siu SWI, Vácha R, Jungwirth P, et al. (2008) Biomolecular simulations of membranes: Physical properties from different force fields. J Chem Phys 128: 125103. doi: 10.1063/1.2897760
|
| [23] | Schrödinger LLC (2010) The PyMOL Molecular Graphics System, Version 1.3r1. |
| [24] |
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun 91: 43–56. doi: 10.1016/0010-4655(95)00042-E
|
| [25] | Lindahl E, Hess B, van der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7: 306–317. |
| [26] |
van der Spoel D, Lindahl E, Hess B, et al. (2005) GROMACS: Fast, flexible, and free. J Comput Chem 26: 1701–1718. doi: 10.1002/jcc.20291
|
| [27] |
Hess B, Kutzner C, van der Spoel D, et al. (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4: 435–447. doi: 10.1021/ct700301q
|
| [28] | Pronk S, Páll S, Schulz R, et al. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29: 845–854. |
| [29] |
Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. J Mol Graph 14: 33–38. doi: 10.1016/0263-7855(96)00018-5
|
| [30] |
Hornak V, Abel R, Okur A, et al. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65: 712–725. doi: 10.1002/prot.21123
|
| [31] | Cornell WD, Cieplak P, Bayly CI, et al. (1995) A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc: 5179–5197. |
| [32] |
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N ⋅ log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089–10092. doi: 10.1063/1.464397
|
| [33] | Hess B, Bekker H, Berendsen HJC, et al. (1997) LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18: 1463–1472. |
| [34] |
Wang J, Wolf RM, Caldwell JW, et al. (2004) Development and testing of a general amber force field. J Comput Chem 25: 1157–1174. doi: 10.1002/jcc.20035
|
| [35] |
Kabsch W, and Sander C (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. doi: 10.1002/bip.360221211
|
| [36] |
Holm L, Sander C (1992) Evaluation of protein models by atomic solvation preference. J Mol Biol 225: 93–105. doi: 10.1016/0022-2836(92)91028-N
|
| [37] |
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8: 127–134. doi: 10.1093/protein/8.2.127
|
| [38] |
Laskowski RA, Swindells MB (2011) LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J Chem Inf Model 51: 2778–2786. doi: 10.1021/ci200227u
|
| [39] | Buck DK, Collins AA (2004) POV-Ray – The Persistence of Vision Raytracer. |
| [40] |
Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Method Neurosci 25: 366–428. doi: 10.1016/S1043-9471(05)80049-7
|
| [41] |
Apweiler R, Bairoch A, Wu CH, et al. (2004) UniProt: the Universal Protein Knowledgebase. Nucleic Acids Res 32: 115–119. doi: 10.1093/nar/gkh151
|
| [42] |
Feng Z, Alqarni MH, Yang P, et al. (2014) Modeling, molecular dynamics simulation, and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J Chem Inf Model 54: 2483–2499. doi: 10.1021/ci5002718
|
| [43] |
Kralj A, Kurt E, Tschammer N, et al. (2014) Synthesis and Biological Evaluation of Biphenyl Amides That Modulate the US28 Receptor. ChemMedChem 9: 151–168. doi: 10.1002/cmdc.201300369
|
| [44] | Rodriguez D, Gutiérrez-de-Terán H (2012) Characterization of the homodimerization interface and functional hotspots of the CXCR4 chemokine receptor. Proteins 80: 1919–1928. |
| [45] |
Dror RO, Arlow DH, Maragakis P, et al. (2011) Activation mechanism of the beta2-adrenergic receptor. Proc Natl Acad Sci U S A 108: 18684–18689. doi: 10.1073/pnas.1110499108
|
| [46] |
Rosenbaum DM, Zhang C, Lyons JA, et al. (2011) Structure and function of an irreversible agonist-beta2 adrenoceptor complex. Nature 469: 236–240. doi: 10.1038/nature09665
|
| [47] | Deupi X, Kobilka B (2007) Activation of G Protein-Coupled Receptors. Mechanisms and Pathways of Heterotrimeric G Protein Signaling. Academic Press, 137–166. |
| [48] |
Lodowski DT, Angel TE, Palczewski K (2009) Comparative Analysis of GPCR Crystal Structures. Photochem Photobiol 85: 425–430. doi: 10.1111/j.1751-1097.2008.00516.x
|
| [49] |
Venkatakrishnan AJ, Deupi X, Lebon G, et al. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494: 185–194. doi: 10.1038/nature11896
|
| [50] |
Tehan BG, Bortolato A, Blaney FE, et al. (2014) Unifying family A GPCR theories of activation. Pharmacol Ther 143: 51–60. doi: 10.1016/j.pharmthera.2014.02.004
|
| [51] |
Yohannan S, Faham S, Yang D, et al. (2004) The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc Natl Acad Sci U S A 101: 959–963. doi: 10.1073/pnas.0306077101
|
| [52] |
Angel TE, Chance MR, Palczewski K (2009) Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc Natl Acad Sci U S A 106: 8555–8560. doi: 10.1073/pnas.0903545106
|
| [53] |
Angel TE, Gupta S, Jastrzebska B, et al. (2009) Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A 106: 14367–14372. doi: 10.1073/pnas.0901074106
|
| [54] |
Piirainen H, Ashok Y, Nanekar RT, et al. (2011) Structural features of adenosine receptors: From crystal to function. Biochim Biophys Acta - Biomembranes 1808: 1233–1244. doi: 10.1016/j.bbamem.2010.05.021
|
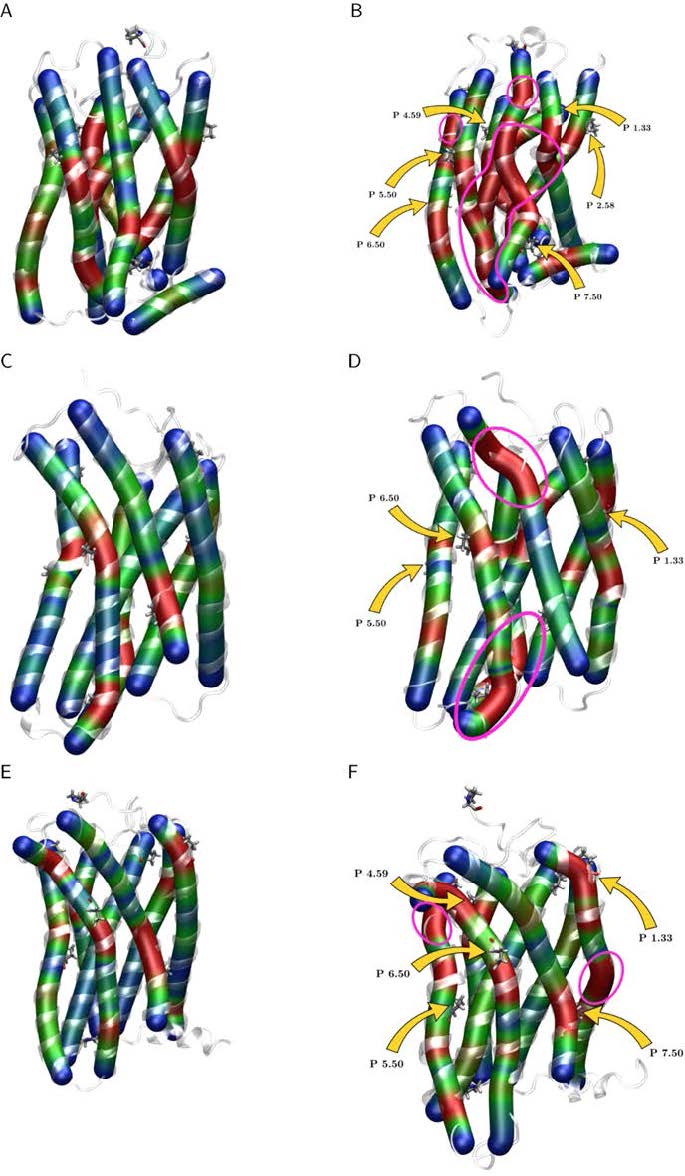
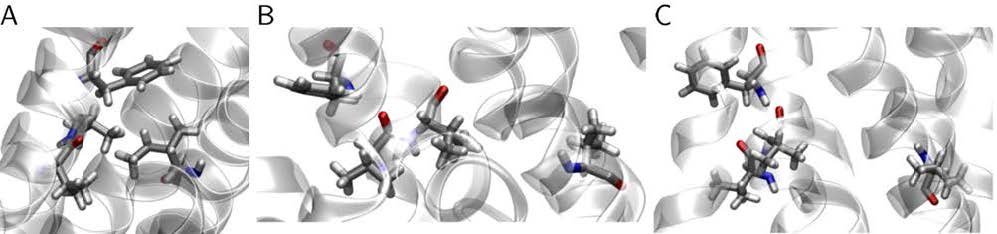
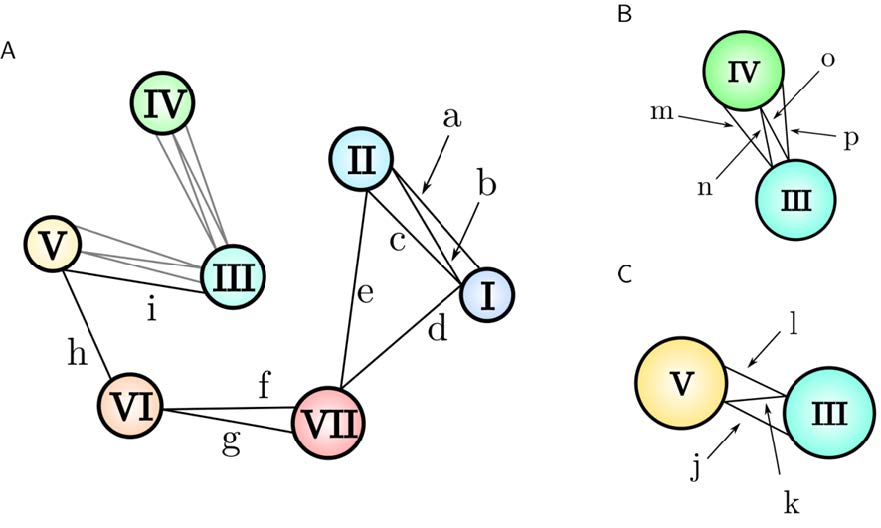
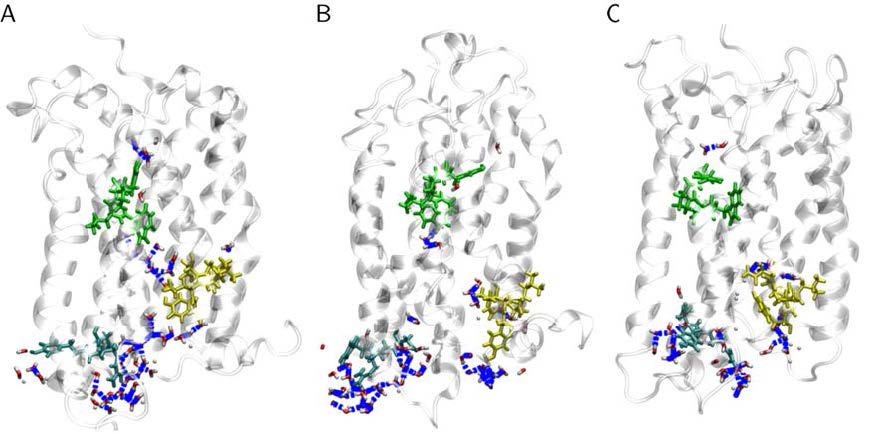

Figures(15) / Tables(3)

Anna Kahler, Heinrich Sticht. A modeling strategy for G-protein coupled receptors[J]. AIMS Biophysics, 2016, 3(2): 211-231. doi: 10.3934/biophy.2016.2.211







 DownLoad:
DownLoad: