Citation: Daniel J. McGrail, Krishan S. Patel, Niti N. Khambhati, Kishan Pithadia, Michelle R. Dawson. Utilizing temporal variations in chemotherapeutic response to improve breast cancer treatment efficacy[J]. AIMS Bioengineering, 2015, 2(4): 310-323. doi: 10.3934/bioeng.2015.4.310

| [1] |
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277-300. doi: 10.3322/caac.20073

|
| [2] |
Thomas E, Holmes FA, Smith TL, et al. (2004) The use of alternate, non-cross-resistant adjuvant chemotherapy on the basis of pathologic response to a neoadjuvant doxorubicin-based regimen in women with operable breast cancer: Long-term results from a prospective randomized trial. J Clin Oncol 22: 2294-2302. doi: 10.1200/JCO.2004.05.207
|
| [3] |
Perola E (2010) An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J Med Chem 53: 2986-2997. doi: 10.1021/jm100118x
|
| [4] |
Macarron R, Banks MN, Bojanic D, et al. (2011) Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 10: 188-195. doi: 10.1038/nrd3368
|
| [5] |
Yu M, Bardia A, Aceto N, et al. (2014) Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345: 216-220. doi: 10.1126/science.1253533
|
| [6] |
Kunz-Schughart LA, Freyer JP, Hofstaedter F, et al. (2004) The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J Biomol Screen 9: 273-285. doi: 10.1177/1087057104265040
|
| [7] |
Kepp O, Galluzzi L, Lipinski M, et al. (2011) Cell death assays for drug discovery. Nat Rev Drug Discov 10: 221-237. doi: 10.1038/nrd3373
|
| [8] |
Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. (2007) Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol 608: 1-22. doi: 10.1007/978-0-387-74039-3_1
|
| [9] | McGrail DJ, Khambhati NN, Qi MX, et al. (2015) Alterations in Ovarian Cancer Cell Adhesion Drive Taxol Resistance by Increasing Microtubule Dynamics in a FAK-dependent Manner. Sci Rep 5: 1-11. |
| [10] |
Hall MD, Telma KA, Chang K-E, et al. (2014) Say No to DMSO: Dimethylsulfoxide Inactivates Cisplatin, Carboplatin, and Other Platinum Complexes. Cancer Res 74: 3913-3922. doi: 10.1158/0008-5472.CAN-14-0247
|
| [11] |
Shoemaker RH, Shoemaker RH (2006) The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6: 813-823. doi: 10.1038/nrc1951
|
| [12] |
Vichai V, Kirtikara K (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1: 1112-1116. doi: 10.1038/nprot.2006.179
|
| [13] |
Monks A, Scudiero D, Skehan P, et al. (1991) Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 83: 757-766. doi: 10.1093/jnci/83.11.757
|
| [14] |
Minotti G, Menna P, Salvatorelli E (2004) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56: 185-229. doi: 10.1124/pr.56.2.6
|
| [15] | Benjamin RS, Riggs CE. Jr, Bachur NR (1977) Plasma Pharmacokinetics of Adriamycin and Its Metabolites in Humans with Normal Hepatic and Renal Function. Cancer Res 37: 1416-1420. |
| [16] | Heggie GD, Sommadossi JP, Cross DS, et al. (1987) Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res 47: 2203-2206. |
| [17] | Den Hartigh J, McVie JG, van Oort WJ, et al. (1983) Pharmacokinetics of mitomycin C in humans. Cancer Res 43: 5017-5021. |
| [18] |
Gaver RC, Colombo N, Green MD, et al. (1988) The disposition of carboplatin in ovarian cancer patients. Cancer Chemother Pharmacol 22: 263-270. doi: 10.1007/BF00273422
|
| [19] | Mok TS, Kanekal S, Lin XR, et al. (2001) Pharmacokinetic study of intralesional cisplatin for the treatment of hepatocellular carcinoma. Cancer 91: 2369-2377. |
| [20] | Longnecker SM, Donehower RC, Cates AE, et al. (1987) High-performance liquid chromatographic assay for taxol in human plasma and urine and pharmacokinetics in a phase I trial. Cancer Treat Rep 71: 53-59. |
| [21] |
Nelson RL (1982) The comparative clinical pharmacology and pharmacokinetics of vindesine, vincristine, and vinblastine in human patients with cancer. Med Pediatr Oncol 10: 115-127. doi: 10.1002/mpo.2950100202
|
| [22] | Au JLS, Li D, Gan Y, et al. (1998) Pharmacodynamics of immediate and delayed effects of paclitaxel: Role of slow apoptosis and intracellular drug retention. Cancer Res 58: 2141-2148. |
| [23] | Zasadil LM, Andersen KA, Yeum D, et al. (2014) Cytotoxicity of Paclitaxel in Breast Cancer Is due to Chromosome Missegregation on Multipolar Spindles. Sci Transl Med 6: 229ra43-229ra43. |
| [24] |
Kern W, Braess J, Friedrichsen S, et al. (2001) Carboplatin pharmacokinetics in patients receiving carboplatin and paclitaxel/docetaxel for advanced lung cancers: impact of age and renal function on area under the curve. J Cancer Res Clin Oncol 127: 64-68. doi: 10.1007/s004320000169
|
| [25] | Cruet-Hennequart S, Villalan S, Kaczmarczyk A, et al. (2009) Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle 8: 3039-3050. |
| [26] |
Wilding JL, Bodmer WF (2014) Cancer Cell Lines for Drug Discovery and Development. Cancer Res 74: 2377-2384. doi: 10.1158/0008-5472.CAN-13-2971
|
| [27] |
Daniel VC, Marchionni L, Hierman JS, et al. (2009) A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res 69: 3364-3373. doi: 10.1158/0008-5472.CAN-08-4210
|
| [28] |
Koshiba H, Hosokawa K, Mori T, et al. (2009) Intravenous paclitaxel is specifically retained in human gynecologic carcinoma tissues in vivo. Int J Gynecol Cancer 19: 484-488. doi: 10.1111/IGC.0b013e3181a130db
|
| [29] |
Peters GJ, Lankelma J, Kok RM, et al. (1993) Prolonged retention of high concentrations of 5-fluorouracil in human and murine tumors as compared with plasma. Cancer Chemother Pharmacol 31: 269-276. doi: 10.1007/BF00685670
|
| [30] |
Laginha KM, Verwoert S, Charrois GJR, et al. (2005) Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin Cancer Res 11: 6944-6949. doi: 10.1158/1078-0432.CCR-05-0343
|
| [31] |
Mori T, Kinoshita Y, Watanabe A, et al. (2006) Retention of paclitaxel in cancer cells for 1 week in vivo and in vitro. Cancer Chemother Pharmacol 58: 665-672. doi: 10.1007/s00280-006-0209-6
|
| [32] |
Bicaku E, Xiong Y, Marchion DC, et al. (2012) In vitro analysis of ovarian cancer response to cisplatin, carboplatin, and paclitaxel identifies common pathways that are also associated with overall patient survival. Br J Cancer 106: 1967-1975. doi: 10.1038/bjc.2012.207
|
| [33] |
Ozols RF, Bundy BN, Greer BE, et al. (2003) Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21: 3194-3200. doi: 10.1200/JCO.2003.02.153
|
| [34] |
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3: 330-338. doi: 10.1038/nrc1074
|
| [35] |
Fallahi-Sichani M, Honarnejad S, Heiser LM, et al. (2013) Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol 9: 708-714. doi: 10.1038/nchembio.1337
|
| [36] |
Haibe-Kains B, El-Hachem N, Birkbak NJ, et al. (2013) Inconsistency in large pharmacogenomic studies. Nature 504: 389-393. doi: 10.1038/nature12831
|
| [37] |
De Souza R, Zahedi P, Badame RM, et al. (2011) Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Mol Cancer Ther 10: 1289-1299. doi: 10.1158/1535-7163.MCT-11-0058
|
| [38] |
Kim PS, Lee PP, Levy D (2008) Dynamics and potential impact of the immune response to chronic myelogenous leukemia. PLoS Comput. Biol 4: e1000095. doi: 10.1371/journal.pcbi.1000095
|
| [39] |
Pertusini E (2001) Investigating the platelet-sparing mechanism of paclitaxel/carboplatin combination chemotherapy. Blood 97: 638-644. doi: 10.1182/blood.V97.3.638
|
| [40] |
Fodale V, Pierobon M, Liotta L, et al. (2011) Mechanism of cell adaptation: when and how do cancer cells develop chemoresistance?. Cancer J 17: 89-95. doi: 10.1097/PPO.0b013e318212dd3d
|
| [41] |
Di Michele M, Della Corte A, Cicchillitti L, et al. (2009) A proteomic approach to paclitaxel chemoresistance in ovarian cancer cell lines. Biochim Biophys Acta 1794: 225-236. doi: 10.1016/j.bbapap.2008.09.017
|
| [42] | Chuthapisith S, Layfield R, Kerr ID, et al. (2007) Proteomic profiling of MCF-7 breast cancer cells with chemoresistance to different types of anti-cancer drugs. Int J Oncol 30: 1545-1551. |
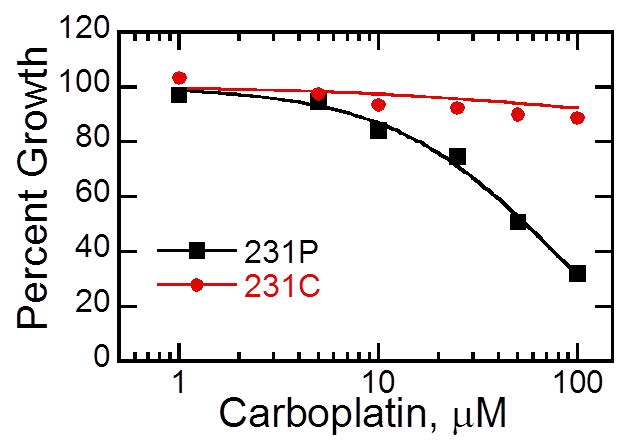
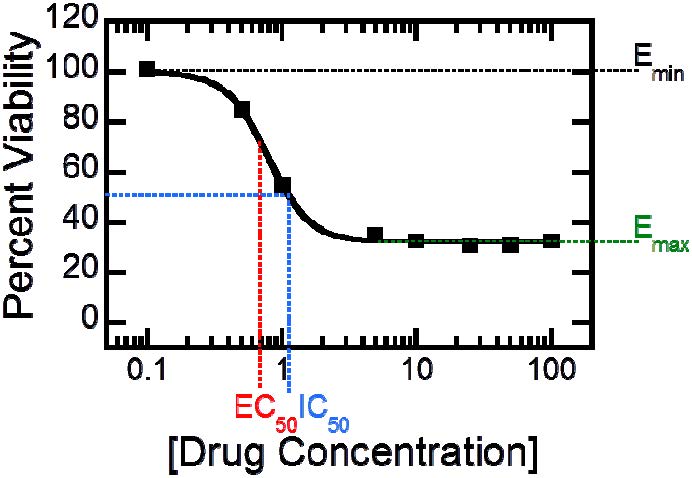
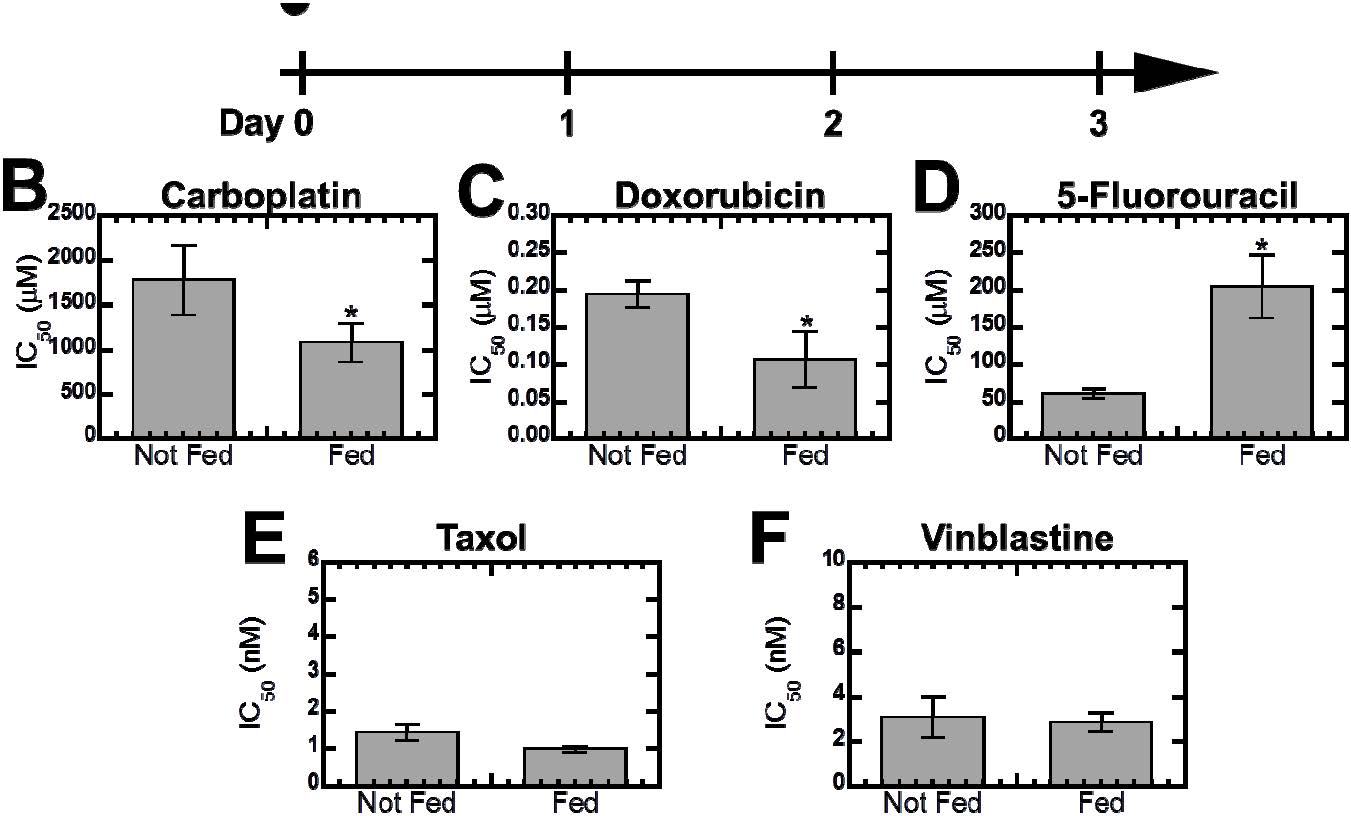
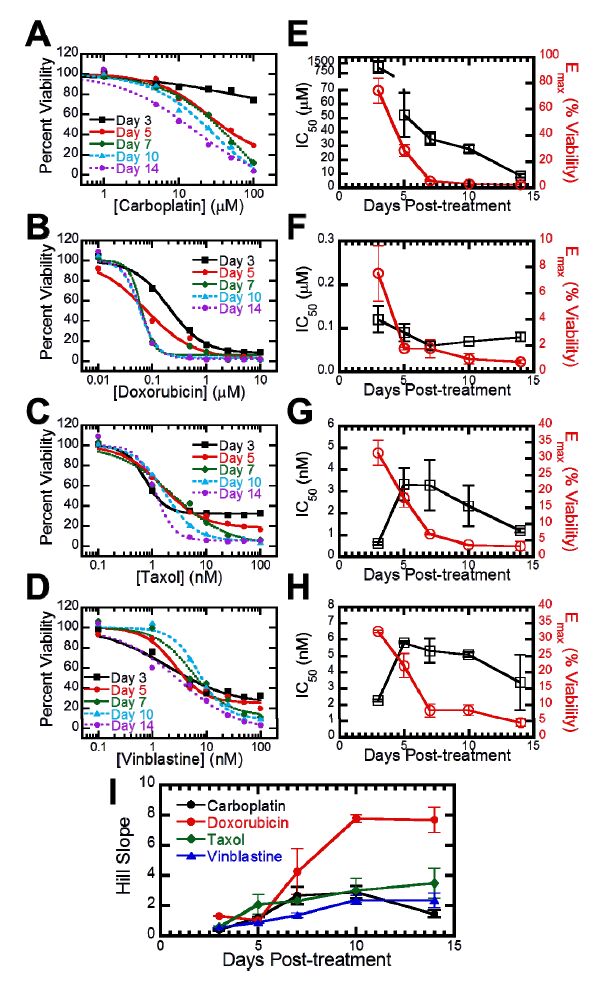
Figures(6) / Tables(1)

Daniel J. McGrail, Krishan S. Patel, Niti N. Khambhati, Kishan Pithadia, Michelle R. Dawson. Utilizing temporal variations in chemotherapeutic response to improve breast cancer treatment efficacy[J]. AIMS Bioengineering, 2015, 2(4): 310-323. doi: 10.3934/bioeng.2015.4.310






 DownLoad:
DownLoad: