Citation: Dongyang Yang, Wei Xu. Statistical modeling on human microbiome sequencing data[J]. Big Data and Information Analytics, 2019, 4(1): 1-12. doi: 10.3934/bdia.2019001

| [1] |
Whiteside SA, Razvi H, Dave S, et al. (2015) The microbiome of the urinary tracta role beyond infection. Nat Rev Urol 12: 81-90. doi: 10.1038/nrurol.2014.361

|
| [2] |
Cho I and Blaser MJ, (2012) The human microbiome: At the interface of health and disease. Nat Rev Genet 13: 260-270. doi: 10.1038/nrg3182
|
| [3] |
HMP Integrative, (2014) The integrative human microbiome project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 16: 276-289. doi: 10.1016/j.chom.2014.08.014
|
| [4] | Young VB, (2017) The role of the microbiome in human health and disease: An introduction for clinicians. BMJ 356: j831. |
| [5] |
Singh RK, Chang HW, Yan D, et al. (2017) Influence of diet on the gut microbiome and implications for human health. J Trans Med e 15: 73. doi: 10.1186/s12967-017-1175-y
|
| [6] |
Hollister EB, Gao C and Versalovic J, (2014) Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 146: 1449-1458. doi: 10.1053/j.gastro.2014.01.052
|
| [7] |
Sampson TR, Debelius JW, Thron T, et al. (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinsons disease. Cell 167: 1469-1480. doi: 10.1016/j.cell.2016.11.018
|
| [8] |
Greenblum S, Turnbaugh PT and Borenstein E, (2012) Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci 109: 594-599. doi: 10.1073/pnas.1116053109
|
| [9] |
Morgan XC, Tickle TL, Sokol H, et al. (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13: R79. doi: 10.1186/gb-2012-13-9-r79
|
| [10] |
Samuel BS and Gordon JI, (2006) A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci 103: 10011-10016. doi: 10.1073/pnas.0602187103
|
| [11] |
Holmes E, Li JV, Athanasiou T, et al. (2011) Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol 19: 349-359. doi: 10.1016/j.tim.2011.05.006
|
| [12] |
Turpin W, Espin-Garcia O, Xu W, et al. (2016) Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 48: 1413-1417. doi: 10.1038/ng.3693
|
| [13] |
Schloissnig S, Arumugam M, Sunagawa S, et al. (2013) Genomic variation landscape of the human gut microbiome. Nature 493: 45-50. doi: 10.1038/nature11711
|
| [14] | Chase J, Fouquier J, Zare M, et al. (2016) Geography and location are the primary drivers of office microbiome composition. mSystems 1: e00022-16. |
| [15] |
Kong HH, Oh J, Deming C, et al. (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22: 850-859. doi: 10.1101/gr.131029.111
|
| [16] |
Grice EA, Kong HH, Conlan S, et al. (2009) Topographical and temporal diversity of the human skin microbiome. Science 324: 1190-1192. doi: 10.1126/science.1171700
|
| [17] |
Turnbaugh PJ, Ley RE, Mahowald MA, et al. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027-1031. doi: 10.1038/nature05414
|
| [18] |
Kau AL, Ahern PP, Griffin NW, et al. (2011) Human nutrition, the gut microbiome and the immune system. Nature 474: 327-336. doi: 10.1038/nature10213
|
| [19] |
Tringe SG and Rubin EM, (2005) Metagenomics: Dna sequencing of environmental samples. Nat Rev Genet 6: 805. doi: 10.1038/nrg1709
|
| [20] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) Qiime allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336. doi: 10.1038/nmeth.f.303
|
| [21] |
Blaxter M, Mann J, Chapman T, et al. (2005) Defining operational taxonomic units using dna barcode data. Philos Trans R Soc, B 360: 1935-1943. doi: 10.1098/rstb.2005.1725
|
| [22] |
Lin W, Shi P, Feng R, et al. (2014) Variable selection in regression with compositional covariates. Biometrika 101: 785-797. doi: 10.1093/biomet/asu031
|
| [23] |
Hongzhe Li, (2015) Microbiome, metagenomics, and high-dimensional compositional data analysis. Annu Rev Stat Its Appl 2: 73-94. doi: 10.1146/annurev-statistics-010814-020351
|
| [24] |
Shankar J, Szpakowski S, Solis NV, et al. (2015) A systematic evaluation of high-dimensional, ensemble-based regression for exploring large model spaces in microbiome analyses. BMC Bioinf 16: 31. doi: 10.1186/s12859-015-0467-6
|
| [25] |
McMurdie PJ and Holmes S, (2014) Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10: e1003531. doi: 10.1371/journal.pcbi.1003531
|
| [26] |
La Rosa PS, Brooks JP, Deych E, et al. (2012) Hypothesis testing and power calculations for taxonomic-based human microbiome data. PloS One 7: e52078. doi: 10.1371/journal.pone.0052078
|
| [27] |
Chen W, Liu F, Ling Z, et al. (2012) Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One 7: e39743. doi: 10.1371/journal.pone.0039743
|
| [28] |
Iwai S, Fei M, Huang D, et al. (2012) Oral and airway microbiota in hiv-infected pneumonia patients. J Clin Microbiol 50: 2995-3002. doi: 10.1128/JCM.00278-12
|
| [29] |
Kim KA, Jung IH, Park SH, et al. (2013) Comparative analysis of the gut microbiota in people with different levels of ginsenoside rb1 degradation to compound k. PLoS One 8: e62409. doi: 10.1371/journal.pone.0062409
|
| [30] |
Xu L, Paterson AD, Turpin W, et al. (2015) Assessment and selection of competing models for zero-inflated microbiome data. PloS One 10: e0129606. doi: 10.1371/journal.pone.0129606
|
| [31] |
Xu L, Paterson AD and Xu W, (2017) Bayesian latent variable models for hierarchical clustered count outcomes with repeated measures in microbiome studies. Genet Epidemiol 41: 221-232. doi: 10.1002/gepi.22031
|
| [32] |
Shestopaloff K, Escobar MD and Xu W, (2018) Analyzing differences between microbiome communities using mixture distributions. Stat Med 37: 4036-4053. doi: 10.1002/sim.7896
|
| [33] | Anderson MJ, (2001) A new method for non-parametric multivariate analysis of variance. Aust Ecol 26: 32-46. |
| [34] |
Jostins L, Ripke S, Weersma RK, et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119-124. doi: 10.1038/nature11582
|
| [35] |
Goodrich JK, Waters JL, Poole AC, et al. (2014) Human genetics shape the gut microbiome. Cell 159: 789-799. doi: 10.1016/j.cell.2014.09.053
|
| [36] |
Blekhman R, Goodrich JK, Huang K, et al. (2015) Host genetic variation impacts microbiome composition across human body sites. Genome Biol 16: 191. doi: 10.1186/s13059-015-0759-1
|
| [37] |
Espin-Garcia O, Croitoru K and Xu W, (2019) A finite mixture model for x-chromosome association with an emphasis on microbiome data analysis. Genet Epidemiol 43: 427-439. doi: 10.1002/gepi.22190
|
| [38] |
David Clayton, (2008) Testing for association on the x chromosome. Biostatistics 9: 593-600. doi: 10.1093/biostatistics/kxn007
|
| [39] |
Zheng G, Joo J, Zhang C, et al. (2007) Testing association for markers on the x chromosome. Genet Epidemiol 31: 834-843. doi: 10.1002/gepi.20244
|
| [40] |
Kevans D, Turpin W, Madsen K, et al. (2015) Determinants of intestinal permeability in healthy first-degree relatives of individuals with crohn's disease. Inflammatory Bowel Dis 21: 879-887. doi: 10.1097/MIB.0000000000000323
|
| [41] |
Matson V, Fessler J, Bao R, et al. (2018) The commensal microbiome is associated with anti-pd-1 efficacy in metastatic melanoma patients. Science 359: 104-108. doi: 10.1126/science.aao3290
|
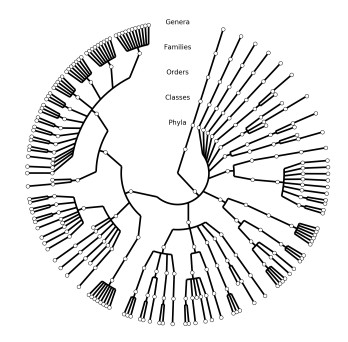
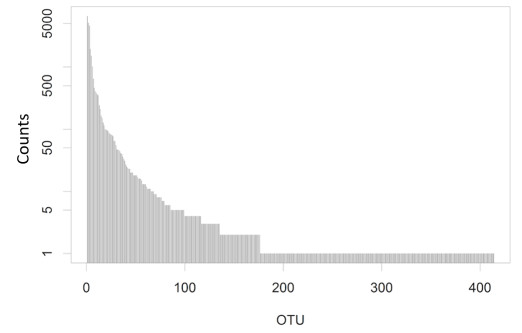
Figures(2)

Dongyang Yang, Wei Xu. Statistical modeling on human microbiome sequencing data[J]. Big Data and Information Analytics, 2019, 4(1): 1-12. doi: 10.3934/bdia.2019001









 DownLoad:
DownLoad: