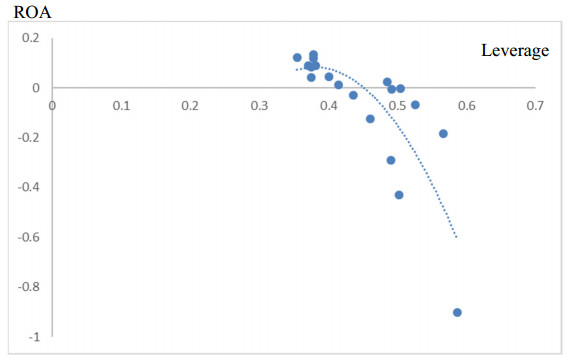
This study analyzes the leverage and performance relationship in the context of the U.S. hospitality industry. We consider that, studying this traditional corporate finance issue in the context of the hospitality industry, is relevant due to its unique characteristics in terms of capital structure and value creation. In addition to Ordinary Least Squares (OLS) and Fixed-Random effects (FE-RE) estimations, we also employ System Generalized Method of Moments (GMM) panel data techniques to avoid the endogeneity issue. Thus, using a sample of 313 U.S. hospitality firms for the period 2001–2018, our primary results are consistent with the pecking order theory, suggesting a negative relationship between leverage and firm performance. The findings are robust to alternative variables description and econometric techniques. We also find an inverted U-shape relationship, but given the high indebtedness of hospitality firms, the negative impact on firm performance is prevalent. Our contribution to the literature is double. First, we highlight the importance of analyzing the capital structure issue in a certain industry and, second, we provide important policy implications for managers and investors.
Citation: Conrado Diego García-Gómez, Mehmet Huseyin Bilgin, Ender Demir, José María Díez-Esteban. Leverage and performance: the case of the U.S. hospitality industry[J]. Quantitative Finance and Economics, 2021, 5(2): 228-246. doi: 10.3934/QFE.2021010
This study analyzes the leverage and performance relationship in the context of the U.S. hospitality industry. We consider that, studying this traditional corporate finance issue in the context of the hospitality industry, is relevant due to its unique characteristics in terms of capital structure and value creation. In addition to Ordinary Least Squares (OLS) and Fixed-Random effects (FE-RE) estimations, we also employ System Generalized Method of Moments (GMM) panel data techniques to avoid the endogeneity issue. Thus, using a sample of 313 U.S. hospitality firms for the period 2001–2018, our primary results are consistent with the pecking order theory, suggesting a negative relationship between leverage and firm performance. The findings are robust to alternative variables description and econometric techniques. We also find an inverted U-shape relationship, but given the high indebtedness of hospitality firms, the negative impact on firm performance is prevalent. Our contribution to the literature is double. First, we highlight the importance of analyzing the capital structure issue in a certain industry and, second, we provide important policy implications for managers and investors.

| [1] | Abdullah H, Tursoy T (2019) Capital structure and firm performance: evidence of Germany under IFRS adoption. Rev Managerial Sci, 1–20. |
| [2] | Ampenberger M, Schmid T, Achleitner AK, et al. (2013) Capital structure decisions in family firms: empirical evidence from a bank-based economy. Rev Managerial Sci 7: 247–275. |
| [3] | Arellano M, Bond S (1991) Some tests of specification for panel data: Monte Carlo evidence and an application to employment equations. Rev Econ Stud 58: 277–297. |
| [4] | Assaf AG, Tsionas MG (2019) Revisiting shape and moderation effects in curvilinear models. Tourism Manage 75: 216–230. |
| [5] | Bloom M, Milkovich GT (1998) Relationships among risk, incentive pay, and organizational performance. Acad Manage J 41: 283–297. |
| [6] | Blundell R, Bond S, Windmeijer F (2000) Estimation in dynamic panel data models: improving on the performance of the standard GMM estimator, In: T, Baltagi, B. Fomby, & B. Carter Hill (Eds.), Nonstationary Panels, Panel Cointegration, and Dynamic Panels, Bingley: Emerald, 53–92. |
| [7] | Brigham EF, Ehrhardt MC (2011) Financial Management: Theory and Practice (13thed), Mason: South-Western Cengage Learning. |
| [8] | Cho MH (1998) Ownership structure, investment, and the corporate value: an empirical analysis. J Financ Econ 47: 103–121. |
| [9] | Dalci I (2018) Impact of financial leverage on profitability of listed manufacturing firms in China. Pacific Account Rev 30: 410–432. |
| [10] | Damodaran A (2019) Cost of Capital by Sector (US). Available from: http://pages.stern.nyu.edu/~adamodar/New_Home_Page/datafile/wacc.htm. |
| [11] | Davydov D (2016) Debt structure and corporate performance in emerging markets. Res Int Bus Financ 382: 299–311. |
| [12] | Demir E, Díez-Esteban JM, García-Gómez CD (2019) The impact of geopolitical risks on cash holdings of hospitality companies: Evidence from emerging countries. J Hospitality Tourism Manage 39: 166–174. |
| [13] | Díez-Esteban JM, García-Gómez CD, López-Iturriaga FJ, et al. (2017) Corporate risk taking, returns and the nature of major shareholders: evidence from prospect theory. Res Int Bus Financ 42: 900–911. |
| [14] | Díez-Esteban JM, Farinha JB, García-Gómez CD (2016) The role of institutional investors in propagating the 2007 financial crisis in Southern Europe. Res Int Bus Financ 38: 439–454. |
| [15] | Flannery MJ, Hankins KW (2013) Estimating dynamic panel models in corporate finance. J Corp Financ 19: 1–19. |
| [16] | Fosu S (2013) Capital structure, product market competition and firm performance: Evidence from South Africa. Q Rev Econ Financ 53: 140–151. |
| [17] | Gleason KC, Mathur LK, Mathur I (2000) The interrelationship between culture, capital structure, and performance: evidence from European retailers. J Bus Res 50: 185–191. |
| [18] | Hair JF, Black WC, Babin BJ, et al. (2006) Multivariate Data Analysis, Upper Saddle River, NJ.: Prentice-Hall. |
| [19] | Hernández PJ (2020) Reassessing the link between firm size and exports. Eurasian Bus Rev, 10: 207–223 |
| [20] | Ibhagui OW, Olokoyo FO (2018) Leverage and firm performance: new evidence on the role of firm size. North Am J Econ Financ 45: 57–92. |
| [21] | Jang S, Tang CH (2009) Simultaneous impacts of international diversification and financial leverage on profitability. J Hospitality Tourism Res 33: 347–368. |
| [22] | Jensen MC, Meckling WH (1976) Theory of the firm: Managerial behavior, agency costs and ownership structure. J Financ Econ 3: 305–360. |
| [23] | Jensen MC (1986) Agency costs of free cash flow, corporate finance, and takeovers. Am Econ Rev 76: 323–329. |
| [24] | Jiang L, Dalbor M (2017) Factors Impacting Capital Expenditures in the Quick Service Restaurant Industry. J Hospitality Financ Manage 25: 90–100. |
| [25] | Jiraporn P, Liu Y (2008) Capital structure, staggered boards, and firm value. Financ Anal J 64: 49–60. |
| [26] | Kraus A, Litzenberger RH (1973) A state‐preference model of optimal financial leverage. J Financ 28: 911–922. |
| [27] | King MR, Santor E (2008) Family values: ownership structure, performance and capital structure of Canadian firms. J Bank Financ 32: 2423–2432. |
| [28] | Le TPV, Phan TBN (2017) Capital structure and firm performance: Empirical evidence from a small transition country. Res Int Bus Financ 42: 710–726. |
| [29] | Li Y, Singal M (2019) Capital structure in the hospitality industry: The role of the asset-light and fee-oriented strategy. Tourism Manage 70: 124–133. |
| [30] | Mao Z, Gu Z (2008) The relationship between financial factors and firm performance: empirical evidence from US restaurant firms. J Foodservice Bus Res 11: 138–159. |
| [31] | Margaritis D, Psillaki M (2010) Capital structure, equity ownership and firm performance. J Bank Financ 34: 621–632. |
| [32] | Modigliani F, Miller MH (1958) The cost of capital, corporation finance and the theory of investment. Am Econ Rev 48: 261–297. |
| [33] | Moudud-Ul-Huq S (2019) Banks' capital buffers, risk, and efficiency in emerging economies: are they counter-cyclical? Eurasian Econ Rev 9: 467–492. |
| [34] | Muradoğlu YG, Sivaprasad S (2014) The impact of leverage on stock returns in the hospitality sector: Evidence from the UK. Tourism Anal 19: 161–171. |
| [35] | Myers SC, Majluf NS (1984) Corporate financing and investment decisions when firms have information that investors do not have. J Financ Econ 13: 187–221. |
| [36] | Nenu EA, Vintilă G, Gherghina SF (2018) The Impact of Capital Structure on Risk and Firm Performance: Empirical Evidence for the Bucharest Stock Exchange Listed Companies. Int J Financ Stud 6: 41. |
| [37] | Pachecho L, Tavares F (2017) Capital structure determinants of hospitality sector SMEs. Tourism Econ 23: 113–132. |
| [38] | Phillips PA, Sipahioglu MA (2004) Performance implications of capital structure: Evidence from quoted UK organizations with hotel interests. Serv Ind J 24: 31–51. |
| [39] | Roberts MR, Whited TM (2013) Endogeneity in empirical corporate finance, In: G. M. Constantinides, M. Harris, & R. M. Stulz (Eds.), Handbook of the Economics of Finance, Elsevier, 493–572. |
| [40] | Roodman D (2008) XTABOND2: Stata module to extend xtabond dynamic panel data estimator, In: Statistical Software Components S435901, Boston College Department of Economics. |
| [41] | Sardo F, Serrasqueiro Z (2021) Intellectual capital and high-tech firms' financing choices in the European context: a panel data analysis. Quant Financ Econ 5: 1–18. |
| [42] | Seo K (2018) Excessive leverage and firm performance in competitive casino markets. Tourism Hospitality Res 18: 498–504. |
| [43] | Singal M (2015) How is the hospitality and tourism industry different? An empirical test of some structural characteristics. Int J Hospitality Manage 47: 116–119. |
| [44] | Sucurro M, Constanzo GD (2019) Ownership structure and firm patenting activity in Italy. Eurasian Econ Rev 9: 239–266. |
| [45] | Tekker D, Tasseven O, Takel A (2009) Determinants of Capital Structure for Turkish Firms: A Panel Data Analysis. Int J Financ Econ 29: 180–187. |
| [46] | Tian GG, Zeitun R (2007) Capital structure and corporate performance: evidence from Jordan. Australasian Account Bus Financ J. |
| [47] | Villarón-Peramato O, Martinez-Ferrero J, García-Sánchez IM (2018) CSR as entrenchment strategy and capital structure: corporate governance and investor protection as complementary and substitutive factors. Rev Managerial Scienc 12: 27–64. |
Figures(4) / Tables(9)

Conrado Diego García-Gómez, Mehmet Huseyin Bilgin, Ender Demir, José María Díez-Esteban. Leverage and performance: the case of the U.S. hospitality industry[J]. Quantitative Finance and Economics, 2021, 5(2): 228-246. doi: 10.3934/QFE.2021010







 DownLoad:
DownLoad: