Citation: Thomas C. Chiang. Economic policy uncertainty and stock returns—evidence from the Japanese market[J]. Quantitative Finance and Economics, 2020, 4(3): 430-458. doi: 10.3934/QFE.2020020

| [1] |
Antonakakis N, Chatziantoniou I, Filis G (2013) Dynamic co-movements of stock market returns, implied volatility and policy uncertainty. Econ Lett 120: 87-92. doi: 10.1016/j.econlet.2013.04.004

|
| [2] |
Aono K, Iwaisako T (2010) On the Predictability of Japanese stock returns using dividend yield. Asia-Pac Financ Mark 17: 141-149. doi: 10.1007/s10690-009-9105-5
|
| [3] | Arbatli EC, Davis SJ, Ito A, et al. (2017) Policy uncertainty in Japan. IMF Working Papers 17/128, International Monetary Fund. |
| [4] |
Arouri M, Estay C, Rault C, et al. (2016) Economic policy uncertainty and stock markets: Long run evidence from the US. Financ Res Lett 18: 136-141. doi: 10.1016/j.frl.2016.04.011
|
| [5] | Bahmani-Oskooee M, Saha S (2019a) On the effects of policy uncertainty on stock prices. J Econ Financ 43: 764-778. |
| [6] | Bahmani-Oskooee M, Saha S (2019b) On the effects of policy uncertainty on stock prices: An asymmetric analysis. Quant Financ Econ 3: 412-424. |
| [7] |
Bae K, Karolyi G, Stulz R (2003). A New Approach to Measuring Financial Contagion. Rev Financ Stud 16: 717-763. doi: 10.1093/rfs/hhg012
|
| [8] |
Baker SR, Bloom N, Davis S (2016) Measuring economic policy uncertainty. Q J Econ 131: 1593-1636. doi: 10.1093/qje/qjw024
|
| [9] |
Bali TG, Brown SJ, Tang Y (2017) Is economic uncertainty priced in the cross-section of stock returns? J Financ Econ 126: 471-489. doi: 10.1016/j.jfineco.2017.09.005
|
| [10] |
Bali TG, Cakici N (2010) World market risk, country-specific risk and expected returns in international stock markets. J Bank Financ 34: 1152-1165. doi: 10.1016/j.jbankfin.2009.11.012
|
| [11] |
Bali TG, Demirtas KO, Levy H (2009) Is there an intertemporal relation between downside risk and expected returns? J Financ Quant Anal 44: 883-909. doi: 10.1017/S0022109009990159
|
| [12] |
Bali TG, Engle RF (2010) The intertemporal capital asset pricing model with dynamic conditional correlations. J Monetary Econ 57: 377-390. doi: 10.1016/j.jmoneco.2010.03.002
|
| [13] | Bali TG, Peng L (2006) Is there a risk-return tradeoff? Evidence from high frequency data. J Appl Econometrics 21: 1169-1198. |
| [14] |
Balli F, Uddin GS, Mudassar H, et al. (2017) Cross-country determinants of economic policy uncertainty spillovers. Econ Lett 156: 179-183. doi: 10.1016/j.econlet.2017.05.016
|
| [15] |
Bekaert G, Harvey CR (1995) Time-varying world market integration. J Financ 50: 403-444. doi: 10.1111/j.1540-6261.1995.tb04790.x
|
| [16] |
Bekaert G, Hoerova M (2016) What do asset prices have to say about risk appetite and uncertainty? J Bank Financ 67: 103-118. doi: 10.1016/j.jbankfin.2015.06.015
|
| [17] |
Bloom N (2009) The impact of uncertainty shocks. Econometrica 77: 623-685. doi: 10.3982/ECTA6248
|
| [18] |
Bloom N (2014) Fluctuations in uncertainty. J Econ Perspect 28: 153-176. doi: 10.1257/jep.28.2.153
|
| [19] | Bollerslev T (2010) Glossary to ARCH (GARCH), in Volatility and Time Series Econometrics: Essays in Honor of Robert Engle, edited by Tim Bollerslev, Jeffrey Russell, and Mark Watson, Oxford University Press, Oxford, UK. |
| [20] |
Bollerslev T, Chou RY, Kroner KF (1992) ARCH modeling in finance: a review of the theory and empirical evidence. J Econometrics 52: 5-59. doi: 10.1016/0304-4076(92)90064-X
|
| [21] |
Brogaard J, Detzel A (2015) The asset pricing implications of government economic policy uncertainty. Manage Sci 61: 3-18. doi: 10.1287/mnsc.2014.2044
|
| [22] |
Caggiano G, Castelnuovo E, Groshenny N (2014) Uncertainty shocks and unemployment dynamics in U.S. recessions. J Monetary Econ 67: 78-92. doi: 10.1016/j.jmoneco.2014.07.006
|
| [23] |
Campbell JY, Hamao Y (1992) Predictable stock returns in the United States and Japan: A study of long‐term capital market integration. J Financ 47: 43-69. doi: 10.1111/j.1540-6261.1992.tb03978.x
|
| [24] | Campbell JY, Lo AW, MacKinlay AC (1997) The Econometrics of Financial Markets, Princeton, NJ: Princeton University Press, 1997. |
| [25] |
Carleton RN, Norton MA, Asmundson GJ (2007) Fearing the unknown: a short version of the intolerance of uncertainty scale. J Anxiety Disord 21: 105-117. doi: 10.1016/j.janxdis.2006.03.014
|
| [26] |
Chen CWS, Chiang TC, So MKP (2003) Asymmetrical reaction to US stock-return news: Evidence from major stock markets based on a double-threshold model. J Econ Bus 55: 487-502. doi: 10.1016/S0148-6195(03)00051-1
|
| [27] |
Chen CYH, Chiang TC (2016) Empirical analysis of the intertemporal relation between downside risk and expected returns: Evidence from time-varying transition probability models. Eur Financ Manage 22: 749-796. doi: 10.1111/eufm.12079
|
| [28] |
Chen CYH, Chiang TC, Härdle WK (2018) Downside risk and stock returns in the G7 countries: An empirical analysis of their long-run and short-run dynamics. J Bank Financ 93: 21-32. doi: 10.1016/j.jbankfin.2018.05.012
|
| [29] |
Chen J, Jiang F, Tong G (2017) Economic policy uncertainty in China and stock market expected returns. Account Financ 57: 1265-1286. doi: 10.1111/acfi.12338
|
| [30] | Chiang TC (2019a) Economic policy uncertainty, risk and stock returns: Evidence from G7 stock markets. Financ Res Lett 29: 41-49. |
| [31] | Chiang TC (2019b) Financial risk, uncertainty and expected returns: evidence from Chinese equity markets. China Financ Rev Int 9: 425-454. |
| [32] |
Chiang TC, Li H, Zheng D (2015) The Intertemporal Return-Risk Relationship: Evidence from international Markets. J Int Financ Mark Inst Money 39: 156-180. doi: 10.1016/j.intfin.2015.06.003
|
| [33] |
Chiang TC, Jeon BN, Li H (2007) Dynamic correlation analysis of financial contagion: Evidence from Asian markets. J Int Money Financ 26: 1206-1228. doi: 10.1016/j.jimonfin.2007.06.005
|
| [34] |
Chong TTL, Wong YC, Yan IKM (2008) Linkages of the Japanese stock market. Japan World Economy 20: 601-662. doi: 10.1016/j.japwor.2007.06.004
|
| [35] |
Christou C, Cunado J, Gupta R, et al. (2017) Economic policy uncertainty and stock market returns in Pacific-Rim countries: Evidence based on a Bayesian panel VAR model. J Multinal Financ Manage 40: 92-102. doi: 10.1016/j.mulfin.2017.03.001
|
| [36] |
Connolly R, Stivers C, Sun L (2005) Stock market uncertainty and the stock-bond return relation. J Financ Quant Anal 40: 161-194. doi: 10.1017/S0022109000001782
|
| [37] | Cornell B (1983) The money supply announcements puzzle: Review and interpretation. Am Econ Rev 73: 644-657. |
| [38] | Connolly RA, Wang FA (1999) Economic news and stock market linkages: Evidence from the U.S., U.K., and Japan. Proceedings of the Second Joint Central Bank Research Conference on Risk Management and Systemic Risk. Available from: https://www.imes.boj.or.jp/cbrc/cbrc-11.pdf. |
| [39] |
Cornish EA, Fisher RA (1937) Moments and cumulants in the specification of distribution. Rev Int Stat Institute 5: 307-320. doi: 10.2307/1400905
|
| [40] | Davis S (2016) An index of global economic policy uncertainty. NBER Working Paper 22740. Available from: http://faculty.chicagobooth.edu/steven.davis/pdf/GlobalEconomic.pdf. |
| [41] |
Demir E, Gozgor G, Lau CKM, et al. (2018) Does economic policy uncertainty predict the Bitcoin returns? An empirical investigation. Financ Res Lett 26: 145-149. doi: 10.1016/j.frl.2018.01.005
|
| [42] |
Diebold FX, Yilmaz K (2009) Measuring financial asset return and volatility spillovers, with application to global equity markets. Econ J 119: 158-171. doi: 10.1111/j.1468-0297.2008.02208.x
|
| [43] |
Dugas MJ, Ladouceur R (2000) Treatment of Gad: Targeting Intolerance of Uncertainty in Two Types of Worry. Behav Modif 24: 635-657. doi: 10.1177/0145445500245002
|
| [44] |
Edwards S (1982) Exchange rates and 'news': A multi-currency approach. J Int Money Financ 1: 211-224. doi: 10.1016/0261-5606(82)90016-X
|
| [45] | Engle RF (2009) Anticipating correlations: A new paradigm for risk management, Princeton: Princeton University Press. |
| [46] |
Engle RF, Granger CWJ (1987) Co-integration and error correction: Representation, estimation, and testing. Econometrica 55: 251-276. doi: 10.2307/1913236
|
| [47] |
Fernandez-Villaverde J, Guerron-Quintana P, Kuester K, et al. (2015) Fiscal volatility shocks and economic activity. Am Econ Rev 105: 3352-3384. doi: 10.1257/aer.20121236
|
| [48] | Forbes K, Kristin F (2012) The "Big C": Identifying and mitigating contagion. Federal Reserve Bank of Kansas City. Proc-Econ Policy Symposium-Jackson Hole, 23-87. |
| [49] |
French K, Schwert W, Stambaugh R (1987) Expected stock returns and volatility. J Financ Econ 19: 3-29. doi: 10.1016/0304-405X(87)90026-2
|
| [50] |
Ghysels E, Santa-Clara R, Valkanov R (2005) There is a risk-return tradeoff after all. J Financ Econ 76: 509-548. doi: 10.1016/j.jfineco.2004.03.008
|
| [51] |
Glosten L, Jagannathan R, Runkle D (1993) On the relation between the expected value and volatility of the nominal excess return on stocks. J Financ 48: 1779-1801. doi: 10.1111/j.1540-6261.1993.tb05128.x
|
| [52] | Gu M, Sun M, Wu Y, et al. (2018) Economic policy uncertainty and momentum, presented at the 26th Annual Conference on Pacific Basin Finance, Economics, Accounting, and Management Conference, September 6-7, 2018, Rutgers University, USA. |
| [53] |
Guo H, Whitelaw R (2006) Uncovering the risk-return relation in the stock market. J Financ 61: 1433-1463. doi: 10.1111/j.1540-6261.2006.00877.x
|
| [54] |
Hamao Y (2018) Japanese financial market research: A survey. Asia-Pac J Financ Stud 47: 361-380. doi: 10.1111/ajfs.12214
|
| [55] |
Hamao Y, Masulis RW, Ng V (1990) Correlations in price changes and volatility across international stock markets. Rev Financ Stud 3: 281-307. doi: 10.1093/rfs/3.2.281
|
| [56] | Hakkio CS, Keeton WR (2009) Financial stress: What is it, how can it be measured, and why does it matter? Econ Rev 94: 5-50. |
| [57] |
Hansen LP, Sargent TJ, Tallarini TD (1999) Robust permanent income and pricing. Rev Econ Stud 66: 873- 907. doi: 10.1111/1467-937X.00112
|
| [58] |
Harvey CR, Liechty JC, Liechty MW, et al. (2010) Portfolio selection with higher moments. Quant Financ 10: 469-485. doi: 10.1080/14697681003756877
|
| [59] |
Hillen MA, Gutheil CM, Strout TD, et al. (2017) Tolerance of uncertainty: Conceptual analysis, integrative model, and implications for healthcare. Social Sci Med 180: 62-75. doi: 10.1016/j.socscimed.2017.03.024
|
| [60] |
Izadi S, Hassan MK (2018) Portfolio and hedging effectiveness of financial assets of the G7 countries. Eurasian Econ Rev 8: 183-213. doi: 10.1007/s40822-017-0090-0
|
| [61] | Johannsen BK (2014) When are the Effects of Fiscal Policy Uncertainty Large? Finance and Economics Discussion Series 2014-40, Board of Governors of the Federal Reserve System, US. |
| [62] | Karolyi GA, Stulz RM (1996) Why do markets move together? An investigation of US-Japanese stock return comovements. J Financ 51: 951-986. |
| [63] | Kirange DK, Deshmukh RR (2016) Sentiment Analysis of news headlines for stock price prediction. Available from: https://www.researchgate.net/publication/299536363_Sentiment_Analysis_of_ |
| [64] | News_Headlines_for_Stock_Price_Prediction. |
| [65] | 64. Kennedy P (2008) A Guide to Econometrics, 6th ed., Oxford, U.K., Blackwell Publishing. |
| [66] |
65. Klößner S, Sekkel R (2014) International spillovers of policy uncertainty. Econ Lett 124: 508-512. doi: 10.1016/j.econlet.2014.07.015
|
| [67] | 66. Knight F (1921) Risk, Uncertainty, and Profit, 5th ed., New York, Dover Publications. |
| [68] |
67. Koutmos G (2014) Positive feedback trading: A review. Rev Behav Financ 6: 155-162. doi: 10.1108/RBF-08-2014-0043
|
| [69] | 68. Lettau M, Ludvigson SC (2010) Measuring and modeling variation in the risk-return trade-off, In: Aït-Sahalia, Y., Hansen, L., Scheinkman, J.A (Eds.), Handbook of Financial Econometrics, North Holland, Amsterdam. |
| [70] |
69. Li Q, Yang J, Hsiao C, et al. (2005) The relationship between stock returns and volatility in international stock markets. J Empir Financ 12: 650-665. doi: 10.1016/j.jempfin.2005.03.001
|
| [71] |
70. Li X (2017) New evidence on economic policy uncertainty and equity premium. Pacific-Basin Financ J 46: 41-56. doi: 10.1016/j.pacfin.2017.08.005
|
| [72] |
71. Li X, Balcilar M, Gupta R, et al. (2015) The causal relationship between economic policy uncertainty and stock returns in China and India: evidence from a bootstrap rolling-window approach. Emerg Mark Financ Trade 52: 674-689. doi: 10.1080/1540496X.2014.998564
|
| [73] | 72. Li Z, Zhong J (2019) Impact of economic policy uncertainty shocks on China's financial conditions. Financ Res Lett. |
| [74] |
73. Liu L, Zhang T (2015) Economic policy uncertainty and stock market volatility. Financ Res Lett 15: 99-105. doi: 10.1016/j.frl.2015.08.009
|
| [75] |
74. Menzly L, Santos T, Veronesi P (2004) Understanding predictability. J Political Economy 112: 1-47. doi: 10.1086/379934
|
| [76] |
75. Merton RC (1980) On estimating the expected return on the market: An exploratory investigation. J Financ Econ 8: 323-361. doi: 10.1016/0304-405X(80)90007-0
|
| [77] | 76. Mishkin FS (1982) Monetary policy and short‐term interest Rates: An efficient markets‐rational expectations approach. J Financ 37: 63-72. |
| [78] |
77. Nelson D (1991) Conditional heteroskedasticity in asset returns: A new approach. Econometrica 59: 347-370. doi: 10.2307/2938260
|
| [79] |
78. Newey W, West KD (1987) A simple, positive definite, heteroskedasticity and autocorrelation consistent covariance matrix. Econometrica 55: 703-708. doi: 10.2307/1913610
|
| [80] |
79. Ozoguz A (2009) Good times or bad times? Investors' uncertainty and stock returns. Rev Financ Stud 22: 4377-4422. doi: 10.1093/rfs/hhn097
|
| [81] |
80. Pastor L, Veronesi P (2013) Political uncertainty and risk premia. J Financ Econ 110: 520-545. doi: 10.1016/j.jfineco.2013.08.007
|
| [82] |
81. Pearce DK, Roley VV (1983) The reaction of stock prices to unanticipated changes in money: A note. J Financ 38: 1323-1333. doi: 10.1111/j.1540-6261.1983.tb02303.x
|
| [83] |
82. Peng L, Xiong W (2006) Investor attention, overconfidence and category learning. J Financ Econ 80: 563-602. doi: 10.1016/j.jfineco.2005.05.003
|
| [84] |
83. Phan DHB, Sharma SS, Tran VT (2018) Can economic policy uncertainty predict stock returns? Global evidence. J Int Financ Mark Inst Money 55: 134-150. doi: 10.1016/j.intfin.2018.04.004
|
| [85] |
84. Rapach DE, Strauss JK, Zhou G (2013) International stock return predictability: What is the role of the United States? J Financ 68: 1633-1622. doi: 10.1111/jofi.12041
|
| [86] |
85. Roy AD (1952) Safety first and the holding of assets. Econometrica 20: 431-449. doi: 10.2307/1907413
|
| [87] |
86. Scruggs JF (1998) Resolving the puzzling intertemporal relation between the market risk premium and conditional market variance: A two-factor approach. J Financ 53: 575-603. doi: 10.1111/0022-1082.235793
|
| [88] | 87. Sum V (2012) How do stock markets in China and Japan respond to economic policy uncertainty in the United States? Available from: https://ssrn.com/abstract=2092346. |
| [89] |
88. Tetlock P, Saar-Tsechansky M, Macskassy S (2008) More than words: quantifying language to measure firms' fundamentals. J Financ 63: 1437-1467. doi: 10.1111/j.1540-6261.2008.01362.x
|
| [90] |
89. Trung NB (2019) The spillover effect of the US uncertainty on emerging economies: A Panel VAR Approach. Appl Econ Lett 26: 210-216. doi: 10.1080/13504851.2018.1458183
|
| [91] |
90. Tsai IC (2017) The source of global stock market risk: A viewpoint of economic policy uncertainty. Econ Model 60: 122-131. doi: 10.1016/j.econmod.2016.09.002
|
| [92] | 91. Wang GJ, Xie C, Wen D, et al. (2019) When Bitcoin meets economic policy uncertainty (EPU): Measuring risk spillover effect from EPU to Bitcoin. Financ Res Lett 31(C). |
| [93] |
92. Whaley RE (2009) Understanding the VIX. J Portf Manage 35: 98-105. doi: 10.3905/JPM.2009.35.3.098
|
| [94] |
93. Zhang Y (2018) China, Japan and the US stock markets and global financial crisis. Asia-Pac Financ Mark 25 23-45. doi: 10.1007/s10690-018-9237-6
|
 QFE-04-03-020-s001.pdf QFE-04-03-020-s001.pdf |
 |
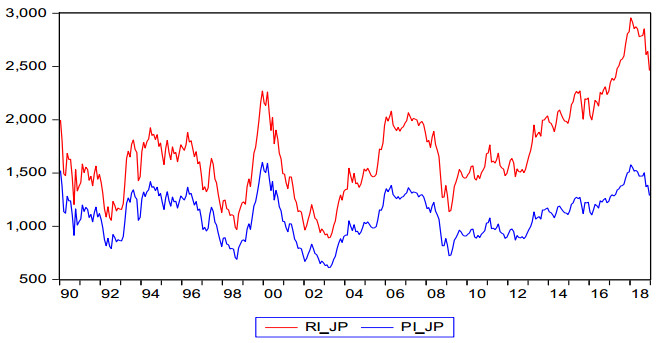
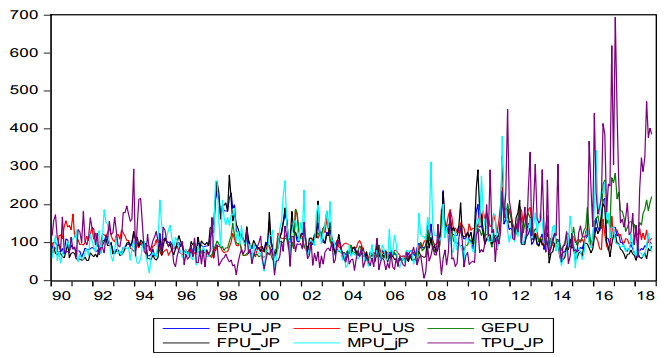
Figures(2) / Tables(8)

Thomas C. Chiang. Economic policy uncertainty and stock returns—evidence from the Japanese market[J]. Quantitative Finance and Economics, 2020, 4(3): 430-458. doi: 10.3934/QFE.2020020







 DownLoad:
DownLoad: