Citation: Manami Inagaki, Masayuki Somei, Tatsunori Oguchi, Ran Ono, Sachie Fukutaka, Ikumi Matsuoka, Mayumi Tsuji, Katsuji Oguchi. Neuroprotective Effects of Dexmedetomidine against Thapsigargin-induced ER-stress via Activity of α2-adrenoceptors and Imidazoline Receptors[J]. AIMS Neuroscience, 2016, 3(2): 237-252. doi: 10.3934/Neuroscience.2016.2.237

| [1] |
Ramsay MA, Luterman DL (2004) Dexmedetomidine as a total intravenous anesthetic agent. Anesthesiology 101: 787-790. doi: 10.1097/00000542-200409000-00028

|
| [2] |
Jalowiecki P, Rudner R, Gonciarz M, et al. (2005) Sole use of dexmedetomidine has limited utility for conscious sedation during outpatient colonoscopy. Anesthesiology 103: 269-273. doi: 10.1097/00000542-200508000-00009
|
| [3] |
Martin E, Ramsay G, Mantz J, et al. (2003) The role of the alpha2-adrenoceptor agonist dexmedetomidine in postsurgical sedation in the intensive care unit. J Intensive Care Med 18: 29-41. doi: 10.1177/0885066602239122
|
| [4] |
Kamibayashi T, Maze M (2000) Clinical uses of alpha2 -adrenergic agonists. Anesthesiology 93: 1345-1349. doi: 10.1097/00000542-200011000-00030
|
| [5] |
Walker SM, Howard RF, Keay KA, et al. (2005) Developmental age influences the effect of epidural dexmedetomidine on inflammatory hyperalgesia in rat pups. Anesthesiology 102: 1226-1234. doi: 10.1097/00000542-200506000-00024
|
| [6] | Hoffman WE, Kochs E, Werner C, et al. (1991) Dexmedetomidine improves neurologic outcome from incomplete ischemia in the rat. Reversal by the alpha 2-adrenergic antagonist atipamezole. Anesthesiology 75: 328-332. |
| [7] |
Dahmani S, Rouelle D, Gressens P, et al. (2010) Characterization of the postconditioning effect of dexmedetomidine in mouse organotypic hippocampal slice cultures exposed to oxygen and glucose deprivation. Anesthesiology 112: 373-383. doi: 10.1097/ALN.0b013e3181ca6982
|
| [8] | Engelhard K, Werner C, Eberspacher E, et al. (2003) The effect of the alpha 2-agonist dexmedetomidine and the N-methyl-D-aspartate antagonist S(+)-ketamine on the expression of apoptosis-regulating proteins after incomplete cerebral ischemia and reperfusion in rats. Anesthesia Analgesia 96: 524-531, table of contents. |
| [9] |
Sato K, Kimura T, Nishikawa T, et al. (2010) Neuroprotective effects of a combination of dexmedetomidine and hypothermia after incomplete cerebral ischemia in rats. Acta anaesthesiologica Scandinavica 54: 377-382. doi: 10.1111/j.1399-6576.2009.02139.x
|
| [10] |
Wikberg JE, Uhlen S, Chhajlani V (1991) Medetomidine stereoisomers delineate two closely related subtypes of idazoxan (imidazoline) I-receptors in the guinea pig. Europ J Pharmacol 193: 335-340. doi: 10.1016/0014-2999(91)90148-J
|
| [11] |
Virtanen R, Savola JM, Saano V, et al. (1988) Characterization of the selectivity, specificity and potency of medetomidine as an alpha 2-adrenoceptor agonist. Europ J Pharmacol 150: 9-14. doi: 10.1016/0014-2999(88)90744-3
|
| [12] |
Savola MK, Savola JM (1996) [3H]dexmedetomidine, an alpha 2-adrenoceptor agonist, detects a novel imidazole binding site in adult rat spinal cord. Europ J Pharmacol 306: 315-323. doi: 10.1016/0014-2999(96)00224-5
|
| [13] |
Dahmani S, Paris A, Jannier V, et al. (2008) Dexmedetomidine increases hippocampal phosphorylated extracellular signal-regulated protein kinase 1 and 2 content by an alpha 2-adrenoceptor-independent mechanism: evidence for the involvement of imidazoline I1 receptors. Anesthesiology 108: 457-466. doi: 10.1097/ALN.0b013e318164ca81
|
| [14] |
Kalimo H, Paljarvi L, Vapalahti M (1979) The early ultrastructural alterations in the rabbit cerebral and cerebellar cortex after compression ischaemia. Neuropathol App Neurobiol 5: 211-223. doi: 10.1111/j.1365-2990.1979.tb00620.x
|
| [15] |
Morimoto K, Yanagihara T (1981) Cerebral ischemia in gerbils: polyribosomal function during progression and recovery. Stroke J Cerebral Circulation 12: 105-110. doi: 10.1161/01.STR.12.1.105
|
| [16] | Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23: 876-882. |
| [17] |
Loop T, Dovi-Akue D, Frick M, et al. (2005) Volatile anesthetics induce caspase-dependent, mitochondria-mediated apoptosis in human T lymphocytes in vitro. Anesthesiology 102: 1147-1157. doi: 10.1097/00000542-200506000-00014
|
| [18] | Nath R, Raser KJ, Hajimohammadreza I, et al. (1997) Thapsigargin induces apoptosis in SH-SY5Y neuroblastoma cells and cerebrocortical cultures. Biochem Mol Biol Int 43: 197-205. |
| [19] |
Nakajima A, Tsuji M, Inagaki M, et al. (2014) Neuroprotective effects of propofol on ER stress-mediated apoptosis in neuroblastoma SH-SY5Y cells. Europ J Pharmacol 725: 47-54. doi: 10.1016/j.ejphar.2014.01.003
|
| [20] |
Zhang M, Shan X, Gu L, et al. (2013) Dexmedetomidine causes neuroprotection via astrocytic α2- adrenergic receptor stimulation and HB-EGF release. J Anesthesiology Clinical Sci 2: 6. doi: 10.7243/2049-9752-2-6
|
| [21] |
Zhang F, Ding T, Yu L, et al. (2012) Dexmedetomidine protects against oxygen-glucose deprivation-induced injury through the I2 imidazoline receptor-PI3K/AKT pathway in rat C6 glioma cells. J Pharm Pharmacol 64: 120-127. doi: 10.1111/j.2042-7158.2011.01382.x
|
| [22] | Salvioli S, Maseroli R, Pazienza TL, et al. (1998) Use of flow cytometry as a tool to study mitochondrial membrane potential in isolated, living hepatocytes. Biochemistry (Mosc) 63: 235-238. |
| [23] | Gertler R, Brown HC, Mitchell DH, et al. (2001) Dexmedetomidine: a novel sedative-analgesic agent. Proceedings 14: 13-21. |
| [24] |
Wikberg JE, Uhlen S, Chhajlani V (1991) Medetomidine stereoisomers delineate two closely related subtypes of idazoxan (imidazoline) I-receptors in the guinea pig. Eur J Pharmacol 193: 335-340. doi: 10.1016/0014-2999(91)90148-J
|
| [25] | Lee YS, Silva AJ (2009) The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci 10: 126-140. |
| [26] | Ho AK, Ogiwara T, Chik CL (1996) Thapsigargin modulates agonist-stimulated cyclic AMP responses through cytosolic calcium-dependent and -independent mechanisms in rat pinealocytes. Mol Pharmacol 49: 1104-1112. |
| [27] |
Kishikawa H, Kobayashi K, Takemori K, et al. (2008) The effects of dexmedetomidine on human neutrophil apoptosis. Biomed Res 29: 189-194. doi: 10.2220/biomedres.29.189
|
| [28] |
Paris A, Mantz J, Tonner PH, et al. (2006) The effects of dexmedetomidine on perinatal excitotoxic brain injury are mediated by the alpha2A-adrenoceptor subtype. Anesthesia Analgesia 102: 456-461. doi: 10.1213/01.ane.0000194301.79118.e9
|
| [29] |
Fujita Y, Inoue K, Sakamoto T, et al. (2013) A comparison between dosages and plasma concentrations of dexmedetomidine in clinically ill patients: a prospective, observational, cohort study in Japan. J Intensive Care 1: 15. doi: 10.1186/2052-0492-1-15
|
| [30] | Masayuki Somei MI, Tatsunori Oguchi, Ran Ono, , Sachie Fukutaka MT, Katsuji Oguchi (2016) Combined neuroprotective effects of propofol and dexmedetomidine on endoplasmic reticulum stress-meditated apoptosis in SH-SY5Y cells. Showa Uni J Med Sci 28. |
| [31] |
Tesson F, Limon-Boulez I, Urban P, et al. (1995) Localization of I2-imidazoline binding sites on monoamine oxidases. J Biol Chem 270: 9856-9861. doi: 10.1074/jbc.270.17.9856
|
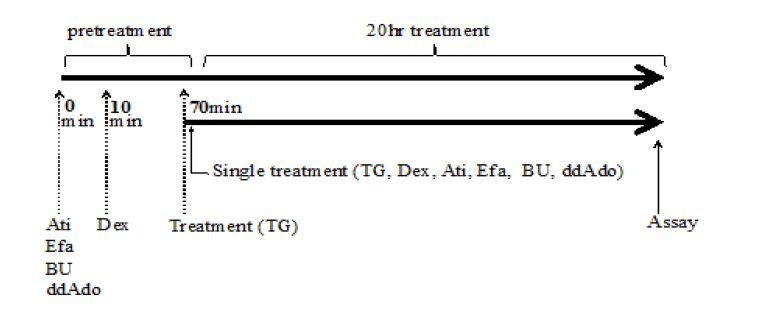
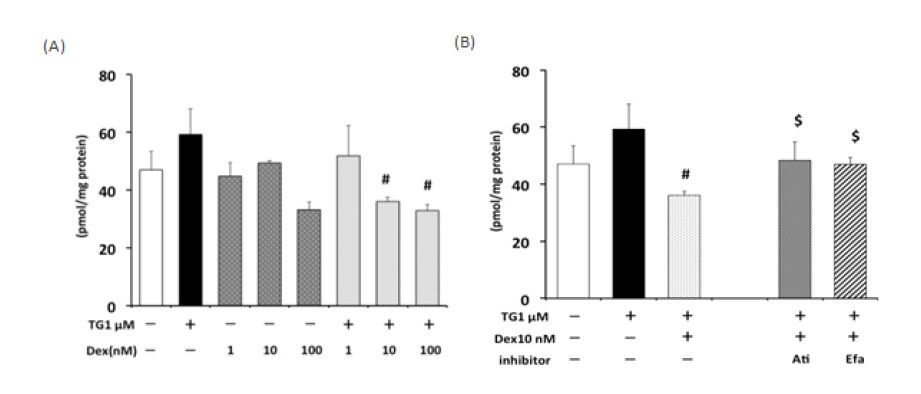
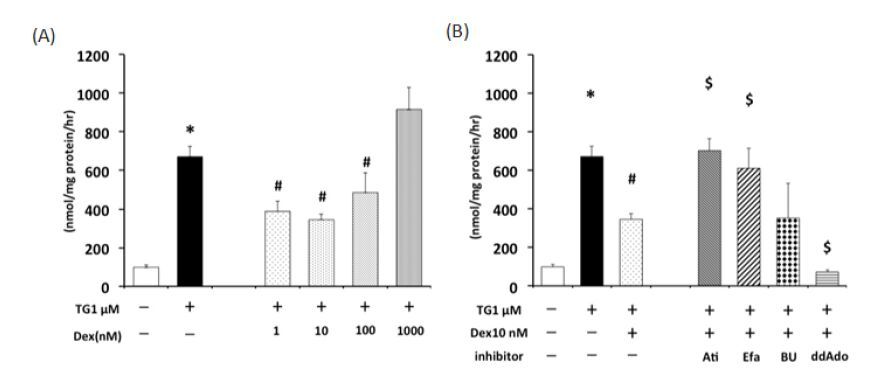
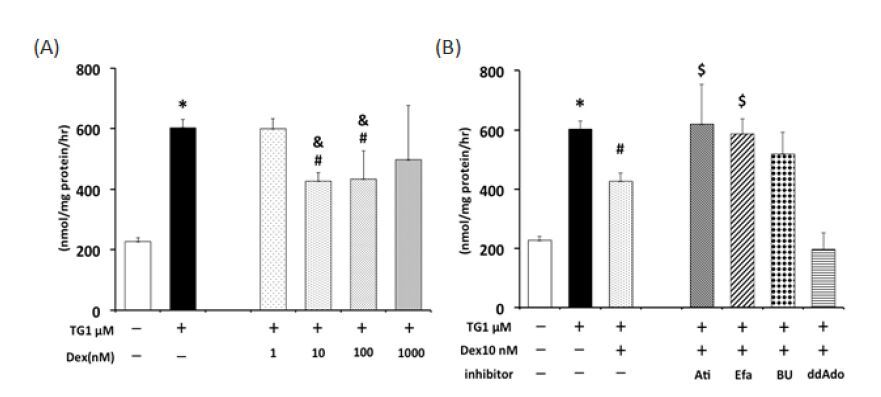
Figures(8)

Manami Inagaki, Masayuki Somei, Tatsunori Oguchi, Ran Ono, Sachie Fukutaka, Ikumi Matsuoka, Mayumi Tsuji, Katsuji Oguchi. Neuroprotective Effects of Dexmedetomidine against Thapsigargin-induced ER-stress via Activity of α2-adrenoceptors and Imidazoline Receptors[J]. AIMS Neuroscience, 2016, 3(2): 237-252. doi: 10.3934/Neuroscience.2016.2.237







 DownLoad:
DownLoad: