Citation: Rafat Zreiq, Souad Kamel, Sahbi Boubaker, Asma A Al-Shammary, Fahad D Algahtani, Fares Alshammari. Generalized Richards model for predicting COVID-19 dynamics in Saudi Arabia based on particle swarm optimization Algorithm[J]. AIMS Public Health, 2020, 7(4): 828-843. doi: 10.3934/publichealth.2020064

| [1] | COVID-19 Worldmeters info website (2020) .Available from: https://www.worldometers.info/coronavirus/. |
| [2] |
Lalwani S, Sahni G, Mewara B, et al. (2020) Predicting optimal lockdown period with parametric approach using three-phase maturation SIRD model for COVID-19 pandemic. Chaos, Solitons Fractals 138: 109939. doi: 10.1016/j.chaos.2020.109939

|
| [3] |
Alboaneen D, Pranggono B, Alshammari D, et al. (2020) Predicting the Epidemiological Outbreak of the Coronavirus Disease 2019 (COVID-19) in Saudi Arabia. Int J Environ Res Public Health 17: 4568. doi: 10.3390/ijerph17124568
|
| [4] |
Almeshal A, Almazrouee A, Alenezi M, et al. (2020) Forecasting the Spread of COVID-19 in Kuwait Using Compartmental and Logistic Regression Models. Appl Sci 10: 3402. doi: 10.3390/app10103402
|
| [5] |
Arora P, Kumar H, Panigrahi P (2020) Prediction and analysis of COVID-19 positive cases using deep learning models: A descriptive case study of India. Chaos, Solitons and Fractals 139: 110017. doi: 10.1016/j.chaos.2020.110017
|
| [6] |
Rodriguez O, Conde-Gutierrez R, Hernadez-Gavier A (2020) Modeling and prediction of COVID-19 in Mexico applying mathematical and computational models. Chaos, Solitons Fractals 138: 109946. doi: 10.1016/j.chaos.2020.109946
|
| [7] |
Hu Z, Cui Q, Han J, et al. (2020) Evaluation and prediction of the COVID-19 variations at different input population and quarantine strategies, a case study in Guangdong province, China. Int J Infect Dis 95: 231-240. doi: 10.1016/j.ijid.2020.04.010
|
| [8] |
Roda W, Varughese M, Han D, et al. (2020) Why is it difficult to accurately predict the COVID-19 epidemic? Infect Dis Modell 5: 271-281. doi: 10.1016/j.idm.2020.03.001
|
| [9] |
Postnikov E (2020) Estimation of COVID-19 dynamics “on a back-of-envelope”: Does the simplest SIR model provide quantitative parameters and predictions? Chaos, Solitons Fractals 135: 109841. doi: 10.1016/j.chaos.2020.109841
|
| [10] |
Ndaïrou F, Area I, Nieto J, et al. (2020) Mathematical modeling of COVID-19 transmission dynamics with a case study of Wuhan. Chaos, Solitons Fractals 135: 109846. doi: 10.1016/j.chaos.2020.109846
|
| [11] |
Alzahrani S, Aljamaan I, Al-fakih E (2020) Forecasting the spread of the COVID-19 pandemic in Saudi Arabia using ARIMA prediction model under current public health interventions. J Infect Public Health 13: 914-919. doi: 10.1016/j.jiph.2020.06.001
|
| [12] |
Aslam M (2020) Using the Kalman filter with Arima for the COVID-19 pandemic dataset of Pakistan. Data Brief 31: 105854. doi: 10.1016/j.dib.2020.105854
|
| [13] | COVID-19 Worldmeters info website (Saudi Arabia) (2020) .Available from: https://www.worldometers.info/coronavirus/country/saudi-arabia/. |
| [14] | COVID-19 Worldmeters info website (Kuwait) (2020) .Available from: https://www.worldometers.info/coronavirus/country/kuwait/. |
| [15] |
Chen D, Chen X, Chen J (2020) Reconstructing and forecasting the COVID-19 epidemic in the United States using a 5-parameter logistic growth model. Global Health Res Policy 5: 25. doi: 10.1186/s41256-020-00152-5
|
| [16] | Malavika B, Marimuth, Joy M, et al. (2020) Forecasting COVID-19 epidemic in India and high incidence states using SIR and logistic growth models. Clin Epidemiol Global Health . |
| [17] |
Munayco C, Tariq A, Rothenberg R, et al. (2020) Early transmission dynamics of COVID-19 in a southern hemisphere setting: Lima-Peru: February 29 the March 30th, 2020. Infect Dise Modell 5: 338-345. doi: 10.1016/j.idm.2020.05.001
|
| [18] |
Wang P, Zheng X, Jiayang L, et al. (2020) Prediction of epidemic trends in COVID-19 with logistic model and machine learning technics. Chaos, Solitons Fractals 139: 110058. doi: 10.1016/j.chaos.2020.110058
|
| [19] |
Behnood A, Gholafshan A, Hosseini S (2020) Determinants of the infection rate of the COVID-19 in the U.S. using ANFIS and virus optimization algorithm (VOA). Chaos, Solitons Fractals 139: 110051. doi: 10.1016/j.chaos.2020.110051
|
| [20] |
Singhal A, Singh P, Lall B, et al. (2020) Modeling and prediction of COVID-19 pandemic using Gaussian mixture model. Chaos, Solitons Fractals 138: 110023. doi: 10.1016/j.chaos.2020.110023
|
| [21] | Wu K, Darcet D, Wang Q, et al. Generalized logistic growth modeling of the COVID-19 outbreak in 29 provinces in China and in the rest of the world (2020) .Available from: https://arxiv.org/abs/2003.05681. |
| [22] |
Shen C (2020) Logistic growth modelling of COVID-19 proliferation in China and its international implications. Int J Infect Dis 96: 582-589. doi: 10.1016/j.ijid.2020.04.085
|
| [23] |
Materassi M (2019) Some fractal thoughts about the COVID-19 infection outbreak. Chaos, Solitons Fractals X4: 100032. doi: 10.1016/j.csfx.2020.100032
|
| [24] |
Al-qaness M, Ewees A, Fan H, et al. (2020) Optimization Method for Forecasting Confirmed Cases of COVID-19 in China. J Clin Med 9: 674. doi: 10.3390/jcm9030674
|
| [25] |
He S, Peng Y, Sun K (2020) SEIR modeling of the COVID-19 and its dynamics. Nonlinear Dyn 101: 1667-1680. doi: 10.1007/s11071-020-05743-y
|
| [26] |
Paggi M (2020) An Analysis of the Italian Lockdown in Retrospective Using Particle Swarm Optimization in Machine Learning Applied to an Epidemiological Model. Physics 2: 368-382. doi: 10.3390/physics2030020
|
| [27] |
Boubaker S (2017) Identification of nonlinear Hammerstein system using mixed integer-real coded particle swarm optimization: application to the electric daily peak-load forecasting. Nonlinear Dyn 90: 797-814. doi: 10.1007/s11071-017-3693-9
|
| [28] | Kennedy J, Eberhart RParticle swarm optimization. (1995) .4: 1942-1948. |
| [29] |
Alberti T, Faranda D (2020) On the uncertainty of real-time predictions of epidemic growths: A COVID-19 case study for China and Italy. Commun Nonlinear Sci Numer Simulat 90: 105372. doi: 10.1016/j.cnsns.2020.105372
|
| [30] |
Consolini G, Materassi M (2020) A stretched logistic equation for pandemic spreading. Chaos, Solitons Fractals 140: 110113. doi: 10.1016/j.chaos.2020.110113
|
| [31] |
Roosa K, Lee Y, Luo R, et al. (2020) Real-time forecasts of the COVID-19 epidemic in China from February 5th to February 24th, 2020. Infect Dis Modell 5: 256-263. doi: 10.1016/j.idm.2020.02.002
|
| [32] | Huang J, Zhang L, Liu X, et al. (2020) Global prediction system for COVID-19 pandemic. Science Bulletin https://doi.org/10.1016/j.scib.2020.08.002. |
| [33] |
Pelinovsky E, Kurkin A, Kurkina O, et al. (2020) Logistic equation and COVID-19. Chaos, Solitons Fractals 140: 110241. doi: 10.1016/j.chaos.2020.110241
|
| [34] | Gillman M, Crokidakis N Dynamics and future of SARS-CoV-2 in the human host (2020) .Available from: https://www.medrxiv.org/content/10.1101/2020.07.14.20153270v2. |
| [35] | Faranda D, Alberti T Modelling the second wave of COVID-19 infections in France and Italy via a Stochastic SEIR model (2020) .Available from: https://arxiv.org/pdf/2006.05081v1.pdf. |
| [36] |
Mwalili S, Kimathi M, Ojiambo V, et al. (2020) SEIR model for COVID-19 dynamics incorporating the environment and social Distancing. BMC Res Notes 13: 352. doi: 10.1186/s13104-020-05192-1
|
| [37] |
Castorina P, Iorio A (2020) Data analysis on Coronavirus spreading by macroscopic growth laws. Int J Mod Phys C 7: 2050103. doi: 10.1142/S012918312050103X
|
| [38] |
Vasconcelos G, Macêdo A, Ospina R, et al. (2020) Modelling fatality curves of COVID-19 and the effectiveness of intervention strategies. Peer J 8: e9421. doi: 10.7717/peerj.9421
|
| [39] |
Maier B, Brockmann D (2020) Effective containment explains subexponential growth in recent confirmed COVID-19 cases in China. Science 368: 742-746. doi: 10.1126/science.abb4557
|
| [40] |
Crokidakis N (2020) COVID-19 spreading in Rio de Janeiro, Brazil: Do the policies of social isolation really work? Chaos, Solitons Fractals 136: 109930. doi: 10.1016/j.chaos.2020.109930
|

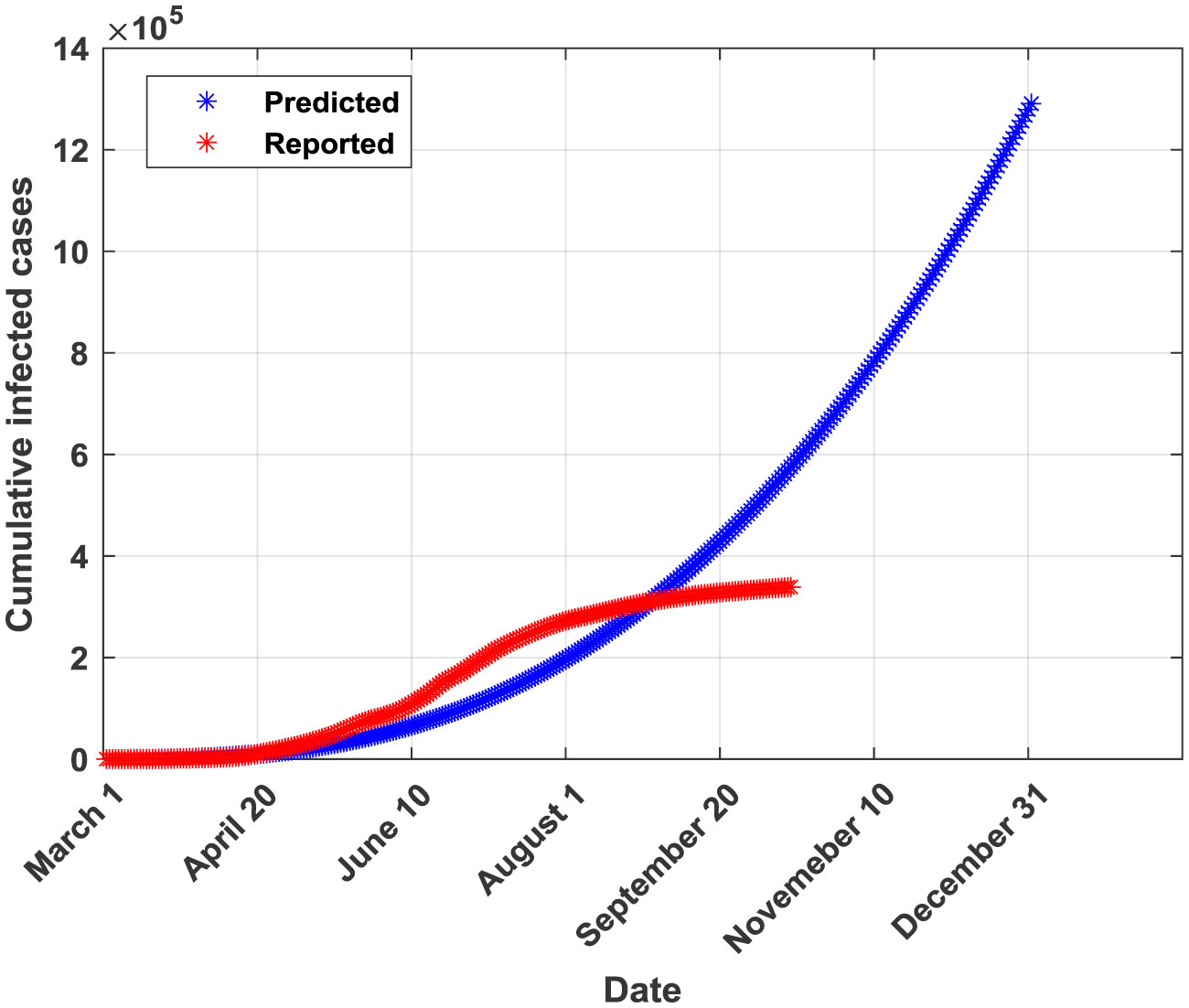

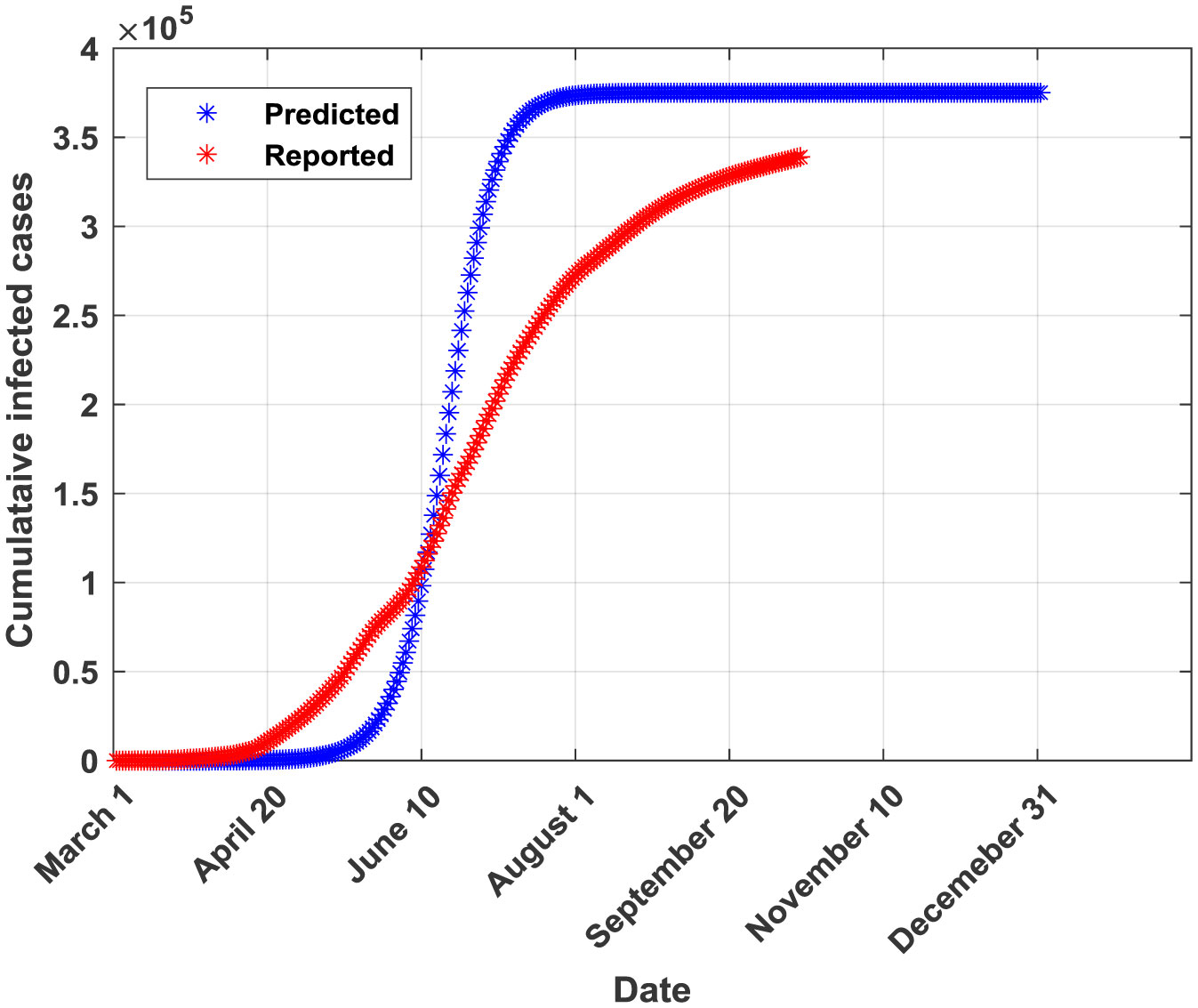
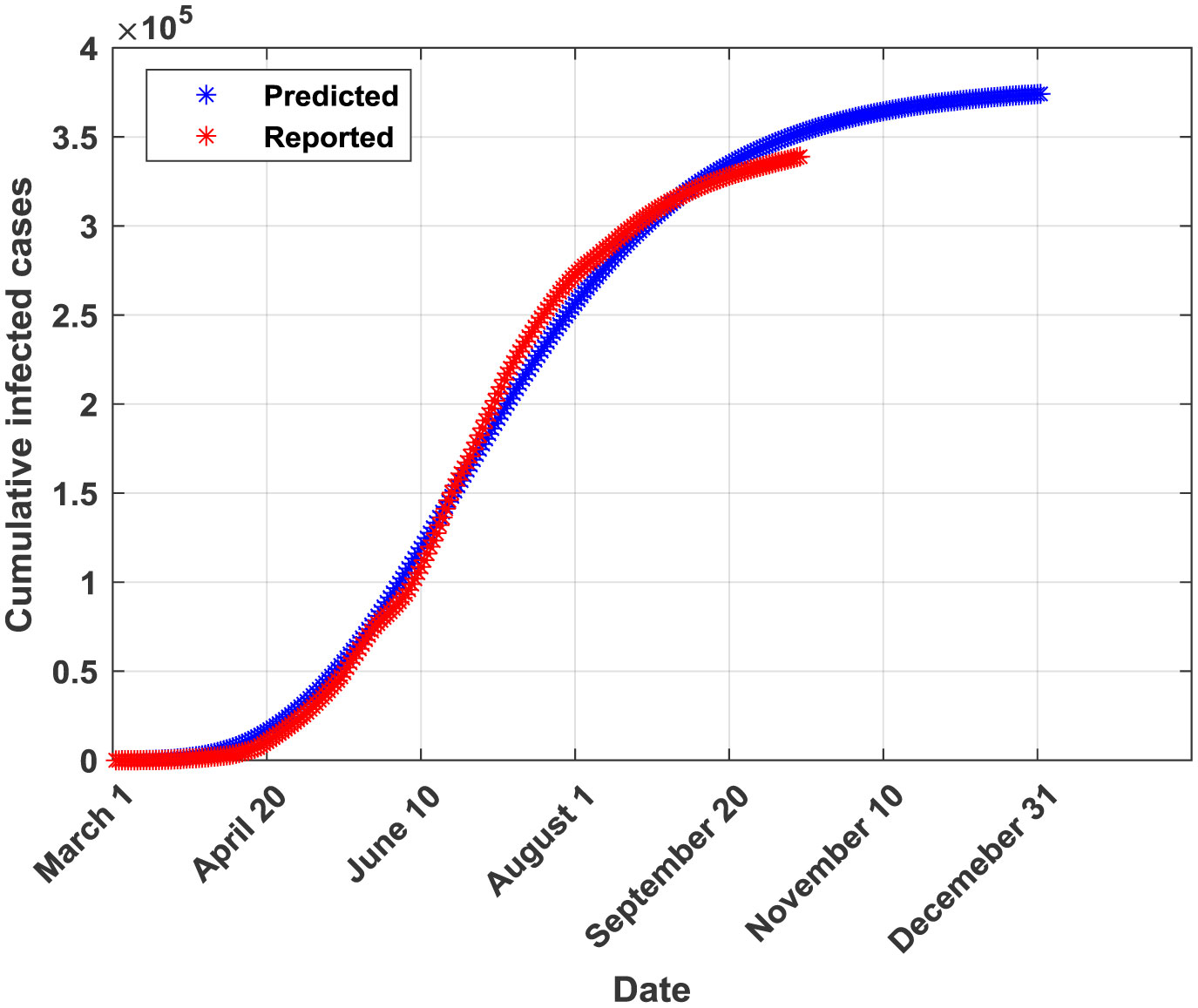
Figures(5) / Tables(6)

Rafat Zreiq, Souad Kamel, Sahbi Boubaker, Asma A Al-Shammary, Fahad D Algahtani, Fares Alshammari. Generalized Richards model for predicting COVID-19 dynamics in Saudi Arabia based on particle swarm optimization Algorithm[J]. AIMS Public Health, 2020, 7(4): 828-843. doi: 10.3934/publichealth.2020064







 DownLoad:
DownLoad: