Citation: Harkirat S. Sethi, Jessica L. Osier, Geordan L. Burks, Jennifer F. Lamar, Hana McFeeters, Robert L. McFeeters. Expedited isolation of natural product peptidyl-tRNA hydrolase inhibitors from a Pth1 affinity column[J]. AIMS Molecular Science, 2017, 4(2): 175-184. doi: 10.3934/molsci.2017.2.175

| [1] |
Lee LA, Puhr ND, Maloney EK, et al. (1994) Increase in antimicrobial-resistant Salmonella infections in the United States, 1989-1990. J Infect Dis 170: 128-134. doi: 10.1093/infdis/170.1.128

|
| [2] |
Glynn MK, Bopp C, Dewitt W, et al. (1998) Emergence of multidrug-resistant Salmonella enterica serotype typhimurium DT104 infections in the United States. New Engl J Med 338: 1333-1338. doi: 10.1056/NEJM199805073381901
|
| [3] | Centers for Disease Control and Prevention: Antibiotic/Antimicrobial Resistance. Centers for Disease Control and Prevention, 2017. Available from: https://www.cdc.gov/drugresistance/ |
| [4] | Kariuki S, Gordon MA, Feasey N, et al. (2015) Antimicrobial resistance and management of invasive Salmonella disease. Vaccine 33 Suppl 3: C21-29. |
| [5] |
Das G, Varshney U (2006) Peptidyl-tRNA hydrolase and its critical role in protein biosynthesis. Microbiology 152: 2191-2195. doi: 10.1099/mic.0.29024-0
|
| [6] | Hernandez-Sanchez J, Valadez JG, Herrera JV, et al. (1998) lambda bar minigene-mediated inhibition of protein synthesis involves accumulation of peptidyl-tRNA and starvation for tRNA. EMBO J 17: 3758-3765. |
| [7] | Cruz-Vera LR, Hernandez-Ramon E, Perez-Zamorano B, et al. (2003) The rate of peptidyl-tRNA dissociation from the ribosome during minigene expression depends on the nature of the last decoding interaction. J Biol Chem 278: 26065-26070. |
| [8] | Tenson T, Herrera JV, Kloss P, et al. (1999) Inhibition of translation and cell growth by minigene expression. J Bacteriol 181: 1617-1622. |
| [9] | Fromant M, Schmitt E, Mechulam Y, et al. (2005) Crystal structure at 1.8 A resolution and identification of active site residues of Sulfolobus solfataricus peptidyl-tRNA hydrolase. Biochemistry 44: 4294-4301. |
| [10] |
Powers R, Mirkovic N, Goldsmith-Fischman S, et al. (2005) Solution structure of Archaeglobus fulgidis peptidyl-tRNA hydrolase (Pth2) provides evidence for an extensive conserved family of Pth2 enzymes in archea, bacteria, and eukaryotes. Protein Sci 14: 2849-2861. doi: 10.1110/ps.051666705
|
| [11] |
Jan Y, Matter M, Pai JT, et al. (2004) A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell 116: 751-762. doi: 10.1016/S0092-8674(04)00204-1
|
| [12] |
Rosas-Sandoval G, Ambrogelly A, Rinehart J, et al. (2002) Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc Natl Acad Scie U S A 99: 16707-16712. doi: 10.1073/pnas.222659199
|
| [13] |
Ito K, Murakami R, Mochizuki M, et al. (2012) Structural basis for the substrate recognition and catalysis of peptidyl-tRNA hydrolase. Nucleic Acids Res 40: 10521-10531. doi: 10.1093/nar/gks790
|
| [14] |
Giorgi L, Bontems F, Fromant M, et al. (2011) RNA-binding site of Escherichia coli peptidyl-tRNA hydrolase. J Biol Chem 286: 39585-39594. doi: 10.1074/jbc.M111.281840
|
| [15] |
Hames MC, McFeeters H, Holloway WB, et al. (2013) Small molecule binding, docking, and characterization of the interaction between Pth1 and peptidyl-tRNA. Int J Mol Sci 14: 22741-22752. doi: 10.3390/ijms141122741
|
| [16] | McFeeters H, Gilbert MJ, Thompson RM, et al. (2012) Inhibition of essential bacterial peptidyl-tRNA hydrolase activity by tropical plant extracts. Nat Prod Commun 7: 1107-1110. |
| [17] |
Kaushik S, Singh N, Yamini S, et al. (2013) The mode of inhibitor binding to peptidyl-tRNA hydrolase: binding studies and structure determination of unbound and bound peptidyl-tRNA hydrolase from Acinetobacter baumannii. PloS One 8: e67547. doi: 10.1371/journal.pone.0067547
|
| [18] |
Giorgi L, Plateau P, O'Mahony G, et al. (2011) NMR-based substrate analog docking to Escherichia coli peptidyl-tRNA hydrolase. J Mol Biol 412: 619-633. doi: 10.1016/j.jmb.2011.06.025
|
| [19] |
Ferguson PP, Holloway WB, Setzer WN, et al. (2016) Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1. Antibiotics 5: 16. doi: 10.3390/antibiotics5020016
|
| [20] |
Kabra A, Shahid S, Pal RK, et al. (2017) Unraveling the stereochemical and dynamic aspects of the catalytic site of bacterial peptidyl-tRNA hydrolase. RNA 23: 202-216. doi: 10.1261/rna.057620.116
|
| [21] |
Goodall JJ, Chen GJ, Page MG (2004) Essential role of histidine 20 in the catalytic mechanism of Escherichia coli peptidyl-tRNA hydrolase. Biochemistry 43: 4583-4591. doi: 10.1021/bi0302200
|
| [22] |
Fromant M, Plateau P, Schmitt E, et al. (1999) Receptor site for the 5'-phosphate of elongator tRNAs governs substrate selection by peptidyl-tRNA hydrolase. Biochemistry 38: 4982-4987. doi: 10.1021/bi982657r
|
| [23] |
Taylor-Creel K, Hames MC, Holloway WB, et al. (2014) Expression, purification, and solubility optimization of peptidyl-tRNA hydrolase 1 from Bacillus cereus. Protein Expr Purif 95: 259-264. doi: 10.1016/j.pep.2014.01.007
|
| [24] | Holloway WB, McFeeters H, Powell AM, et al. (2015) A Highly Adaptable Method for Quantification of Peptidyl-tRNA Hydrolase Activity. J Anal Bioanal Tech 6: 244. |
| [25] | McFeeters H, McFeeters RL (2014) Current Methods for Analysis of Enzymatic Peptidyl-tRNA Hydrolysis. J Anal Bioanal Tech 5: 215. |
| [26] |
El-Elimat T, Raja HA, Day CS, et al. (2017) alpha-Pyrone derivatives, tetra/hexahydroxanthones, and cyclodepsipeptides from two freshwater fungi. Bioorg Med Chem 25: 795-804. doi: 10.1016/j.bmc.2016.11.059
|
| [27] | Harris SM, McFeeters H, Ogungbe IV, et al. (2011) Peptidyl-tRNA hydrolase screening combined with molecular docking reveals the antibiotic potential of Syzygium johnsonii bark extract. Nat Prod Commun 6: 1421-1424. |
| [28] |
Bonin PD, Erickson LA (2002) Development of a fluorescence polarization assay for peptidyl-tRNA hydrolase. Anal Biochem 306: 8-16. doi: 10.1006/abio.2002.5700
|
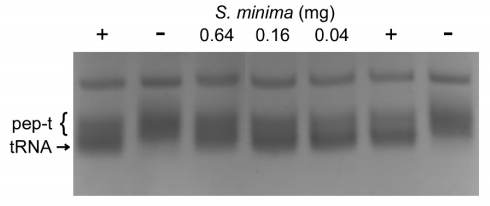
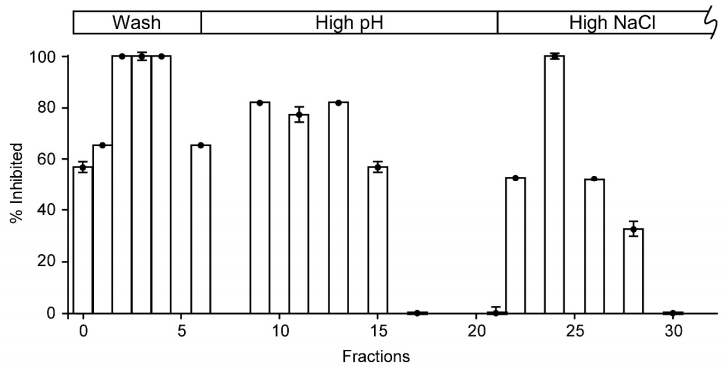
Figures(2)

Harkirat S. Sethi, Jessica L. Osier, Geordan L. Burks, Jennifer F. Lamar, Hana McFeeters, Robert L. McFeeters. Expedited isolation of natural product peptidyl-tRNA hydrolase inhibitors from a Pth1 affinity column[J]. AIMS Molecular Science, 2017, 4(2): 175-184. doi: 10.3934/molsci.2017.2.175







 DownLoad:
DownLoad: