Citation: Sujit Nair. Current insights into the molecular systems pharmacology of lncRNA-miRNA regulatory interactions and implications in cancer translational medicine[J]. AIMS Molecular Science, 2016, 3(2): 104-124. doi: 10.3934/molsci.2016.2.104

| [1] |
Hayes EL, Lewis-Wambi JS (2015) Mechanisms of endocrine resistance in breast cancer: an overview of the proposed roles of noncoding RNA. Breast Cancer Res 17: 40. doi: 10.1186/s13058-015-0542-y

|
| [2] |
Ergun S, Oztuzcu S (2015) Oncocers: ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways. Tumour Biol 36: 3129-3136. doi: 10.1007/s13277-015-3346-x
|
| [3] |
Neelakandan K, Babu P, Nair S (2012) Emerging roles for modulation of microRNA signatures in cancer chemoprevention. Curr Cancer Drug Targets 12: 716-740. doi: 10.2174/156800912801784875
|
| [4] | Li Y, Wang X (2015) Role of long noncoding RNAs in malignant disease (Review). Mol Med Rep 13: 1463-1469. |
| [5] | Lo PK, Wolfson B, Zhou X, et al. (2015) Noncoding RNAs in breast cancer. Brief Funct Genom pii: elv055. |
| [6] | Takayama KI, Inoue S (2015) The emerging role of noncoding RNA in prostate cancer progression and its implication on diagnosis and treatment. Brief Funct Genom pii: elv057. |
| [7] | Xu YJ, Du Y, Fan Y (2015) Long noncoding RNAs in lung cancer: what we know in 2015. Clin Transl Oncol in press. |
| [8] | Xie X, Tang B, Xiao YF, et al. (2015) Long non-coding RNAs in colorectal cancer. Oncotarget 7: 5226-5239. |
| [9] |
Jalali S, Bhartiya D, Lalwani MK, et al. (2013) Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One 8: e53823. doi: 10.1371/journal.pone.0053823
|
| [10] |
Yoon JH, Abdelmohsen K, Gorospe M (2014) Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol 34: 9-14. doi: 10.1016/j.semcdb.2014.05.015
|
| [11] | Cai Y, He J, Zhang D (2015) Long noncoding RNA CCAT2 promotes breast tumor growth by regulating the Wnt signaling pathway. Onco Targets Ther 8: 2657-2664. |
| [12] | Tuo YL, Li XM, Luo J (2015) Long noncoding RNA UCA1 modulates breast cancer cell growth and apoptosis through decreasing tumor suppressive miR-143. Eur Rev Med Pharmacol Sci 19: 3403-3411. |
| [13] |
Wu Q, Guo L, Jiang F, et al. (2015) Analysis of the miRNA-mRNA-lncRNA networks in ER+ and ER- breast cancer cell lines. J Cell Mol Med 19: 2874-2887. doi: 10.1111/jcmm.12681
|
| [14] | Tordonato C, Di Fiore PP, Nicassio F (2015) The role of non-coding RNAs in the regulation of stem cells and progenitors in the normal mammary gland and in breast tumors. Front Genet 6: 72. |
| [15] |
Bailey ST, Westerling T, Brown M (2015) Loss of estrogen-regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer Res 75: 436-445. doi: 10.1158/0008-5472.CAN-14-1041
|
| [16] |
Zhang H, Cai K, Wang J, et al. (2014) MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 32: 2858-2868. doi: 10.1002/stem.1795
|
| [17] |
Li JT, Wang LF, Zhao YL, et al. (2014) Nuclear factor of activated T cells 5 maintained by Hotair suppression of miR-568 upregulates S100 calcium binding protein A4 to promote breast cancer metastasis. Breast Cancer Res 16: 454. doi: 10.1186/s13058-014-0454-2
|
| [18] |
Hou P, Zhao Y, Li Z, et al. (2014) LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis 5: e1287. doi: 10.1038/cddis.2014.249
|
| [19] |
Zhang Z, Zhu Z, Watabe K, et al. (2013) Negative regulation of lncRNA GAS5 by miR-21. Cell Death Differ 20: 1558-1568. doi: 10.1038/cdd.2013.110
|
| [20] |
Augoff K, McCue B, Plow EF, et al. (2012) miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer 11: 5. doi: 10.1186/1476-4598-11-5
|
| [21] |
Matouk IJ, Raveh E, Abu-lail R, et al. (2014) Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta 1843: 1414-1426. doi: 10.1016/j.bbamcr.2014.03.023
|
| [22] |
Paci P, Colombo T, Farina L (2014) Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst Biol 8: 83. doi: 10.1186/1752-0509-8-83
|
| [23] |
Li N, Zhou P, Zheng J, et al. (2014) A polymorphism rs12325489C>T in the lincRNA-ENST00000515084 exon was found to modulate breast cancer risk via GWAS-based association analyses. PLoS One 9: e98251. doi: 10.1371/journal.pone.0098251
|
| [24] |
Zhang J, Fan D, Jian Z, et al. (2015) Cancer Specific Long Noncoding RNAs Show Differential Expression Patterns and Competing Endogenous RNA Potential in Hepatocellular Carcinoma. PLoS One 10: e0141042. doi: 10.1371/journal.pone.0141042
|
| [25] | Li H, Li J, Jia S, et al. (2015) miR675 upregulates long noncoding RNA H19 through activating EGR1 in human liver cancer. Oncotarget 6: 31958-31984. |
| [26] |
Lv J, Ma L, Chen XL, et al. (2014) Downregulation of LncRNAH19 and MiR-675 promotes migration and invasion of human hepatocellular carcinoma cells through AKT/GSK-3beta/Cdc25A signaling pathway. J Huazhong Univ Sci Technol Med Sci 34: 363-369. doi: 10.1007/s11596-014-1284-2
|
| [27] |
Zhang L, Yang F, Yuan JH, et al. (2013) Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 34: 577-586. doi: 10.1093/carcin/bgs381
|
| [28] |
Zamani M, Sadeghizadeh M, Behmanesh M, et al. (2015) Dendrosomal curcumin increases expression of the long non-coding RNA gene MEG3 via up-regulation of epi-miRs in hepatocellular cancer. Phytomedicine 22: 961-967. doi: 10.1016/j.phymed.2015.05.071
|
| [29] |
Wang J, Liu X, Wu H, et al. (2010) CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res 38: 5366-5383. doi: 10.1093/nar/gkq285
|
| [30] |
Cui M, Xiao Z, Wang Y, et al. (2015) Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res 75: 846-857. doi: 10.1158/0008-5472.CAN-14-1192
|
| [31] |
Li T, Xie J, Shen C, et al. (2015) Amplification of Long Noncoding RNA ZFAS1 Promotes Metastasis in Hepatocellular Carcinoma. Cancer Res 75: 3181-3191. doi: 10.1158/0008-5472.CAN-14-3721
|
| [32] |
Cao C, Sun J, Zhang D, et al. (2015) The long intergenic noncoding RNA UFC1, a target of MicroRNA 34a, interacts with the mRNA stabilizing protein HuR to increase levels of beta-catenin in HCC cells. Gastroenterology 148: 415-426 e418. doi: 10.1053/j.gastro.2014.10.012
|
| [33] |
Zhao Q, Li T, Qi J, et al. (2014) The miR-545/374a cluster encoded in the Ftx lncRNA is overexpressed in HBV-related hepatocellular carcinoma and promotes tumorigenesis and tumor progression. PLoS One 9: e109782. doi: 10.1371/journal.pone.0109782
|
| [34] |
Chen CL, Tseng YW, Wu JC, et al. (2015) Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials 44: 71-81. doi: 10.1016/j.biomaterials.2014.12.023
|
| [35] |
Braconi C, Kogure T, Valeri N, et al. (2011) microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene 30: 4750-4756. doi: 10.1038/onc.2011.193
|
| [36] |
Yuan SX, Wang J, Yang F, et al. (2016) Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology 63: 499-511. doi: 10.1002/hep.27893
|
| [37] |
Tsang FH, Au SL, Wei L, et al. (2015) Long non-coding RNA HOTTIP is frequently up-regulated in hepatocellular carcinoma and is targeted by tumour suppressive miR-125b. Liver Int 35: 1597-1606. doi: 10.1111/liv.12746
|
| [38] |
Lempiainen H, Couttet P, Bolognani F, et al. (2013) Identification of Dlk1-Dio3 imprinted gene cluster noncoding RNAs as novel candidate biomarkers for liver tumor promotion. Toxicol Sci 131: 375-386. doi: 10.1093/toxsci/kfs303
|
| [39] |
Tang J, Zhuo H, Zhang X, et al. (2014) A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis 5: e1549. doi: 10.1038/cddis.2014.518
|
| [40] |
Takahashi K, Yan IK, Kogure T, et al. (2014) Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio 4: 458-467. doi: 10.1016/j.fob.2014.04.007
|
| [41] |
Takahashi K, Yan IK, Haga H, et al. (2014) Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J Cell Sci 127: 1585-1594. doi: 10.1242/jcs.141069
|
| [42] |
Yuan JH, Yang F, Wang F, et al. (2014) A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25: 666-681. doi: 10.1016/j.ccr.2014.03.010
|
| [43] | Li DF, Yang MF, Shi SL, et al. (2015) TM4SF5-CTD-2354A18.1-miR-4697-3P may play a key role in the pathogenesis of gastric cancer. Bratisl Lek Listy 116: 608-615. |
| [44] | Xia T, Liao Q, Jiang X, et al. (2014) Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci Rep 4: 6088. |
| [45] |
Zhang ZX, Liu ZQ, Jiang B, et al. (2015) BRAF activated non-coding RNA (BANCR) promoting gastric cancer cells proliferation via regulation of NF-kappaB1. Biochem Biophys Res Commun 465: 225-231. doi: 10.1016/j.bbrc.2015.07.158
|
| [46] |
Song B, Guan Z, Liu F, et al. (2015) Long non-coding RNA HOTAIR promotes HLA-G expression via inhibiting miR-152 in gastric cancer cells. Biochem Biophys Res Commun 464: 807-813. doi: 10.1016/j.bbrc.2015.07.040
|
| [47] |
Liu XH, Sun M, Nie FQ, et al. (2014) Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer 13: 92. doi: 10.1186/1476-4598-13-92
|
| [48] |
Qi P, Xu MD, Shen XH, et al. (2015) Reciprocal repression between TUSC7 and miR-23b in gastric cancer. Int J Cancer 137: 1269-1278. doi: 10.1002/ijc.29516
|
| [49] |
Zhou X, Ye F, Yin C, et al. (2015) The Interaction Between MiR-141 and lncRNA-H19 in Regulating Cell Proliferation and Migration in Gastric Cancer. Cell Physiol Biochem 36: 1440-1452. doi: 10.1159/000430309
|
| [50] |
Dey BK, Pfeifer K, Dutta A (2014) The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28: 491-501. doi: 10.1101/gad.234419.113
|
| [51] |
Zhuang M, Gao W, Xu J, et al. (2014) The long non-coding RNA H19-derived miR-675 modulates human gastric cancer cell proliferation by targeting tumor suppressor RUNX1. Biochem Biophys Res Commun 448: 315-322. doi: 10.1016/j.bbrc.2013.12.126
|
| [52] |
Li H, Yu B, Li J, et al. (2014) Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 5: 2318-2329. doi: 10.18632/oncotarget.1913
|
| [53] |
Hu Y, Wang J, Qian J, et al. (2014) Long noncoding RNA GAPLINC regulates CD44-dependent cell invasiveness and associates with poor prognosis of gastric cancer. Cancer Res 74: 6890-6902. doi: 10.1158/0008-5472.CAN-14-0686
|
| [54] |
Iio A, Takagi T, Miki K, et al. (2013) DDX6 post-transcriptionally down-regulates miR-143/145 expression through host gene NCR143/145 in cancer cells. Biochim Biophys Acta 1829: 1102-1110. doi: 10.1016/j.bbagrm.2013.07.010
|
| [55] |
Yan J, Guo X, Xia J, et al. (2014) MiR-148a regulates MEG3 in gastric cancer by targeting DNA methyltransferase 1. Med Oncol 31: 879. doi: 10.1007/s12032-014-0879-6
|
| [56] |
Fan QH, Yu R, Huang WX, et al. (2014) The has-miR-526b binding-site rs8506G>a polymorphism in the lincRNA-NR_024015 exon identified by GWASs predispose to non-cardia gastric cancer risk. PLoS One 9: e90008. doi: 10.1371/journal.pone.0090008
|
| [57] |
Zhang EB, Kong R, Yin DD, et al. (2014) Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 5: 2276-2292. doi: 10.18632/oncotarget.1902
|
| [58] |
Xu C, Shao Y, Xia T, et al. (2014) lncRNA-AC130710 targeting by miR-129-5p is upregulated in gastric cancer and associates with poor prognosis. Tumour Biol 35: 9701-9706. doi: 10.1007/s13277-014-2274-5
|
| [59] |
Lu L, Luo F, Liu Y, et al. (2015) Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol 289: 276-285. doi: 10.1016/j.taap.2015.09.016
|
| [60] |
Shao T, Wu A, Chen J, et al. (2015) Identification of module biomarkers from the dysregulated ceRNA-ceRNA interaction network in lung adenocarcinoma. Mol Biosyst 11: 3048-3058. doi: 10.1039/C5MB00364D
|
| [61] | You J, Zhang Y, Liu B, et al. (2014) MicroRNA-449a inhibits cell growth in lung cancer and regulates long noncoding RNA nuclear enriched abundant transcript 1. Indian J Cancer 51 Suppl 3: e77-81. |
| [62] |
Chamoux A, Berthon P, Laubignat JF (1996) Determination of maximum aerobic velocity by a five minute test with reference to running world records. A theoretical approach. Arch Physiol Biochem 104: 207-211. doi: 10.1076/apab.104.2.207.12877
|
| [63] |
Yang Y, Li H, Hou S, et al. (2013) The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS One 8: e65309. doi: 10.1371/journal.pone.0065309
|
| [64] |
Prensner JR, Chen W, Han S, et al. (2014) The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 16: 900-908. doi: 10.1016/j.neo.2014.09.001
|
| [65] |
He JH, Zhang JZ, Han ZP, et al. (2014) Reciprocal regulation of PCGEM1 and miR-145 promote proliferation of LNCaP prostate cancer cells. J Exp Clin Cancer Res 33: 72. doi: 10.1186/s13046-014-0072-y
|
| [66] |
Zhu M, Chen Q, Liu X, et al. (2014) lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J 281: 3766-3775. doi: 10.1111/febs.12902
|
| [67] |
Chiyomaru T, Yamamura S, Fukuhara S, et al. (2013) Genistein inhibits prostate cancer cell growth by targeting miR-34a and oncogenic HOTAIR. PLoS One 8: e70372. doi: 10.1371/journal.pone.0070372
|
| [68] |
Martinez-Fernandez M, Rubio C, Segovia C, et al. (2015) EZH2 in Bladder Cancer, a Promising Therapeutic Target. Int J Mol Sci 16: 27107-27132. doi: 10.3390/ijms161126000
|
| [69] |
Luo M, Li Z, Wang W, et al. (2013) Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett 333: 213-221. doi: 10.1016/j.canlet.2013.01.033
|
| [70] |
Martinez-Fernandez M, Feber A, Duenas M, et al. (2015) Analysis of the Polycomb-related lncRNAs HOTAIR and ANRIL in bladder cancer. Clin Epigenetics 7: 109. doi: 10.1186/s13148-015-0141-x
|
| [71] |
He W, Cai Q, Sun F, et al. (2013) linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim Biophys Acta 1832: 1528-1537. doi: 10.1016/j.bbadis.2013.05.010
|
| [72] |
Eissa S, Matboli M, Essawy NO, et al. (2015) Integrative functional genetic-epigenetic approach for selecting genes as urine biomarkers for bladder cancer diagnosis. Tumour Biol 36: 9545-9552. doi: 10.1007/s13277-015-3722-6
|
| [73] |
Fu X, Liu Y, Zhuang C, et al. (2015) Synthetic artificial microRNAs targeting UCA1-MALAT1 or c-Myc inhibit malignant phenotypes of bladder cancer cells T24 and 5637. Mol Biosyst 11: 1285-1289. doi: 10.1039/C5MB00127G
|
| [74] |
Wang T, Yuan J, Feng N, et al. (2014) Hsa-miR-1 downregulates long non-coding RNA urothelial cancer associated 1 in bladder cancer. Tumour Biol 35: 10075-10084. doi: 10.1007/s13277-014-2321-2
|
| [75] |
Li Z, Li X, Wu S, et al. (2014) Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci 105: 951-955. doi: 10.1111/cas.12461
|
| [76] |
Han Y, Liu Y, Zhang H, et al. (2013) Hsa-miR-125b suppresses bladder cancer development by down-regulating oncogene SIRT7 and oncogenic long noncoding RNA MALAT1. FEBS Lett 587: 3875-3882. doi: 10.1016/j.febslet.2013.10.023
|
| [77] |
Andrew AS, Marsit CJ, Schned AR, et al. (2015) Expression of tumor suppressive microRNA-34a is associated with a reduced risk of bladder cancer recurrence. Int J Cancer 137: 1158-1166. doi: 10.1002/ijc.29413
|
| [78] | Wang J, Lei ZJ, Guo Y, et al. (2015) miRNA-regulated delivery of lincRNA-p21 suppresses beta-catenin signaling and tumorigenicity of colorectal cancer stem cells. Oncotarget 6: 37852-37870. |
| [79] |
Hunten S, Kaller M, Drepper F, et al. (2015) p53-Regulated Networks of Protein, mRNA, miRNA, and lncRNA Expression Revealed by Integrated Pulsed Stable Isotope Labeling With Amino Acids in Cell Culture (pSILAC) and Next Generation Sequencing (NGS) Analyses. Mol Cell Proteom 14: 2609-2629. doi: 10.1074/mcp.M115.050237
|
| [80] |
Liang WC, Fu WM, Wong CW, et al. (2015) The lncRNA H19 promotes epithelial to mesenchymal transition by functioning as miRNA sponges in colorectal cancer. Oncotarget 6: 22513-22525. doi: 10.18632/oncotarget.4154
|
| [81] |
Tsang WP, Ng EK, Ng SS, et al. (2010) Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 31: 350-358. doi: 10.1093/carcin/bgp181
|
| [82] |
Franklin JL, Rankin CR, Levy S, et al. (2013) Malignant transformation of colonic epithelial cells by a colon-derived long noncoding RNA. Biochem Biophys Res Commun 440: 99-104. doi: 10.1016/j.bbrc.2013.09.040
|
| [83] |
Ling H, Spizzo R, Atlasi Y, et al. (2013) CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res 23: 1446-1461. doi: 10.1101/gr.152942.112
|
| [84] |
Guo G, Kang Q, Zhu X, et al. (2015) A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene 34: 1768-1779. doi: 10.1038/onc.2014.131
|
| [85] |
Emmrich S, Streltsov A, Schmidt F, et al. (2014) LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol Cancer 13: 171. doi: 10.1186/1476-4598-13-171
|
| [86] |
Xing CY, Hu XQ, Xie FY, et al. (2015) Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett 589: 1981-1987. doi: 10.1016/j.febslet.2015.04.061
|
| [87] |
Iosue I, Quaranta R, Masciarelli S, et al. (2013) Argonaute 2 sustains the gene expression program driving human monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis 4: e926. doi: 10.1038/cddis.2013.452
|
| [88] |
Yao Y, Ma J, Xue Y, et al. (2015) Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett 359: 75-86. doi: 10.1016/j.canlet.2014.12.051
|
| [89] | Cui H, Mu Y, Yu L, et al. (2015) Methylation of the miR-126 gene associated with glioma progression. Fam Cancer 15: 317-324. |
| [90] |
Shi Y, Wang Y, Luan W, et al. (2014) Long non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS One 9: e86295. doi: 10.1371/journal.pone.0086295
|
| [91] |
Bian EB, Li J, Xie YS, et al. (2015) LncRNAs: new players in gliomas, with special emphasis on the interaction of lncRNAs With EZH2. J Cell Physiol 230: 496-503. doi: 10.1002/jcp.24549
|
| [92] |
Shi L, Zhang J, Pan T, et al. (2010) MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain Res 1312: 120-126. doi: 10.1016/j.brainres.2009.11.056
|
| [93] |
Wang P, Liu YH, Yao YL, et al. (2015) Long non-coding RNA CASC2 suppresses malignancy in human gliomas by miR-21. Cell Signal 27: 275-282. doi: 10.1016/j.cellsig.2014.11.011
|
| [94] |
Zeng Q, Wang Q, Chen X, et al. (2016) Analysis of lncRNAs expression in UVB-induced stress responses of melanocytes. J Dermatol Sci 81: 53-60. doi: 10.1016/j.jdermsci.2015.10.019
|
| [95] |
Soares MR, Huber J, Rios AF, et al. (2010) Investigation of IGF2/ApaI and H19/RsaI polymorphisms in patients with cutaneous melanoma. Growth Horm IGF Res 20: 295-297. doi: 10.1016/j.ghir.2010.03.006
|
| [96] |
Chiyomaru T, Fukuhara S, Saini S, et al. (2014) Long non-coding RNA HOTAIR is targeted and regulated by miR-141 in human cancer cells. J Biol Chem 289: 12550-12565. doi: 10.1074/jbc.M113.488593
|
| [97] |
Slaby O, Jancovicova J, Lakomy R, et al. (2010) Expression of miRNA-106b in conventional renal cell carcinoma is a potential marker for prediction of early metastasis after nephrectomy. J Exp Clin Cancer Res 29: 90. doi: 10.1186/1756-9966-29-90
|
| [98] | Liu S, Song L, Zeng S, et al. (2015) MALAT1-miR-124-RBG2 axis is involved in growth and invasion of HR-HPV-positive cervical cancer cells. Tumour Biol in press. |
| [99] |
Kwanhian W, Lenze D, Alles J, et al. (2012) MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med 1: 141-155. doi: 10.1002/cam4.29
|
| [100] |
Wang J, Zhou Y, Lu J, et al. (2014) Combined detection of serum exosomal miR-21 and HOTAIR as diagnostic and prognostic biomarkers for laryngeal squamous cell carcinoma. Med Oncol 31: 148. doi: 10.1007/s12032-014-0148-8
|
| [101] |
Ma MZ, Chu BF, Zhang Y, et al. (2015) Long non-coding RNA CCAT1 promotes gallbladder cancer development via negative modulation of miRNA-218-5p. Cell Death Dis 6: e1583. doi: 10.1038/cddis.2014.541
|
| [102] |
Fang D, Yang H, Lin J, et al. (2015) 17beta-estradiol regulates cell proliferation, colony formation, migration, invasion and promotes apoptosis by upregulating miR-9 and thus degrades MALAT-1 in osteosarcoma cell MG-63 in an estrogen receptor-independent manner. Biochem Biophys Res Commun 457: 500-506. doi: 10.1016/j.bbrc.2014.12.114
|
| [103] | Malouf G, Zhang J, Tannir M, et al. (2015) Charting DNA methylation of long non-coding RNA in clear-cell renal cell carcinoma. ASCO Annual Meeting. Abstract Number 432: May 2015. Available at http://meetinglibrary.asco.org/content/141955-141159. |
| [104] | Wang H, Guo R, Hoffmann A, et al. (2015) Effect of long noncoding RNA RUNXOR on the epigenetic regulation of RUNX1 in acute myelocytic leukemia. ASCO Annual Meeting. Abstract Number 7018: May 2015. Available at http://meetinglibrary.asco.org/content/146827-146156. |
| [105] | Hatziapostolou M, Koutsioumpa M, Kottakis F, et al. (2015) Targeting colon cancer metabolism through a long noncoding RNA. ASCO Annual Meeting. Abstract Number 604: May 2015. Available at http://meetinglibrary.asco.org/content/140661-140158. |
| [106] | Crea F, Parolia A, Xue H, et al. (2015) Identification and functional characterization of a long non-coding RNA driving hormone-independent prostate cancer progression. ASCO Annual Meeting. Abstract Number 11087: May 2015. Available at http://meetinglibrary.asco.org/content/150625-150156. |
| [107] | Broto J, Fernandez-Serra A, Duran J, et al. (2015) Prognostic relevance of miRNA let-7e in localized intestinal GIST: A Spanish Group for Research on Sarcoma (GEIS) Study. ASCO Annual Meeting. Abstract Number 10524: May 2015. Available at http://meetinglibrary.asco.org/content/147760-147156. |
| [108] | Ordonez J, Serrano M, García-Puche J, et al. (2015) miRNA21 as a specific marker for the detection of circulating tumor cell. ASCO Annual Meeting. Abstract Number e22025: May 2015. Available at http://meetinglibrary.asco.org/content/150328-150156. |
| [109] | Ling Z, Mao W (2015) Circulating miRNAs as a potential marker for gefitinib sensitivity and correlation with EGFR mutational status in human lung cancers. ASCO Annual Meeting. Abstract Number e18500: May 2015. Available at http://meetinglibrary.asco.org/content/153292-153156. |
| [110] | Mullane S, Werner L, Zhou C, et al. (2015) Predicting outcome in metastatic urothelial cancer (UC) receiving docetaxel (DT): miRNA profiling in pre and post therapy. ASCO Annual Meeting. Abstract Number e15518: May 2015. Available at http://meetinglibrary.asco.org/content/150309-150156. |
| [111] | Yue L, Yao Y, Qiu W, et al. (2015) MiR-141 to confer the docetaxel chemoresistance of breast cancer cells via regulating EIF4E expression. ASCO Annual Meeting. Abstract Number e12007: May 2015. Available at http://meetinglibrary.asco.org/content/149427-149156. |
| [112] | Espin E, Perez-Fidalgo J, Tormo E, et al. (2015) Effect of trastuzumab on the antiproliferative effects of PI3K inhibitors in HER2+ breast cancer cells with de novo resistance to trastuzumab. ASCO Annual Meeting. Abstract Number e11592: May 2015. Available at http://meetinglibrary.asco.org/content/144338-144156. |
| [113] | Nair S, Liew C, Khor TO, et al. (2014) Differential signaling regulatory networks governing hormone refractory prostate cancers. J Chin Pharm Sci 23: 511-524. |
| [114] | Nair S, Liew C, Khor TO, et al. (2015) Elucidation of regulatory interaction networks underlying human prostate adenocarcinoma. J Chin Pharm Sci 24: 12-27. |
| [115] |
Wang F, Lu J, Peng X, et al. (2016) Integrated analysis of microRNA regulatory network in nasopharyngeal carcinoma with deep sequencing. J Exp Clin Cancer Res 35: 17. doi: 10.1186/s13046-016-0292-4
|
| [116] |
Nair S, Kong AN (2015) Architecture of signature miRNA regulatory networks in cancer chemoprevention. Curr Pharmacol Rep 1: 89-101. doi: 10.1007/s40495-014-0014-6
|
| [117] | Liu Y, Zhao M (2016) lnCaNet: pan-cancer co-expression network for human lncRNA and cancer genes. Bioinformatics pii: 017. |
| [118] | Cao L, Xiao PF, Tao YF, et al. (2016) Microarray profiling of bone marrow long non-coding RNA expression in Chinese pediatric acute myeloid leukemia patients. Oncol Rep 35: 757-770. |
| [119] |
Shannon P, Markiel A, Ozier O, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498-2504. doi: 10.1101/gr.1239303
|
| [120] | Nair S (2016) Pharmacometrics and systems pharmacology of immune checkpoint inhibitor nivolumab in cancer translational medicine. Adv Mod Oncol Res 2: 18-31. |
| [121] |
Elling R, Chan J, Fitzgerald KA (2016) Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol 46: 504-512. doi: 10.1002/eji.201444558
|
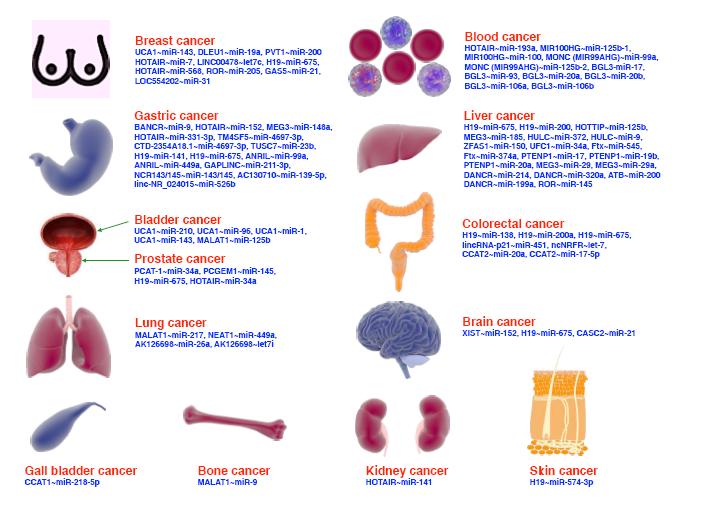
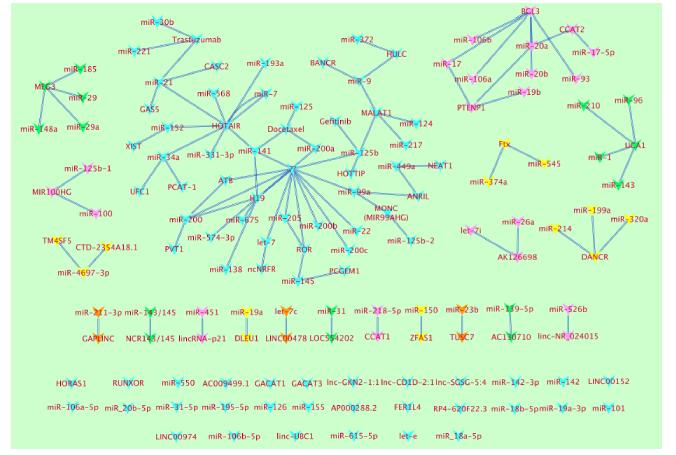
Figures(2)

Sujit Nair. Current insights into the molecular systems pharmacology of lncRNA-miRNA regulatory interactions and implications in cancer translational medicine[J]. AIMS Molecular Science, 2016, 3(2): 104-124. doi: 10.3934/molsci.2016.2.104







 DownLoad:
DownLoad: