Citation: Yuji Hamada, Takeshi Sato, Takashi Taniguchi. Multiscale simulation of a polymer melt flow between two coaxial cylinders under nonisothermal conditions[J]. Mathematics in Engineering, 2021, 3(6): 1-22. doi: 10.3934/mine.2021042

| [1] | Y. Masubuchi, Simulating the flow of entangled polymers, Annu. Rev. Chem. Biomol. Eng., 5 (2014), 11-33. |
| [2] | T. Sato, A review on transport phenomena of polymeric liquids, J. Soc. Rheol. Jpn., 48 (2020), 1-14. |
| [3] | R. B. Bird, R. C. Armstrong, O. Hassager, Dynamics of polymeric liquids, Volume 1 fluid mechanics, 3 Eds., John Wiley & Sons, Inc, 1987. |
| [4] | M. M. Cross, Rheology of non-Newtonian fluids: A new flow equation for pseudoplastic systems, J. Colloid. Sci., 20 (1965), 417-437. |
| [5] | J. D. Ferry, Viscoelastic properties of polymers, 3 Eds., John Wiley & Sons, Inc, 1980. |
| [6] | H. H. Chiang, C. A. Hieber, K. K. Wang, A unified simulation of the filling and postfilling stages in injection molding. Part Ⅰ: Formulation, Polym. Eng. Sci., 31 (1991), 116-124. |
| [7] | R. E. Otmani, M. Zinet, M. Boutaous, H. Benhadid, Numerical simulation and thermal analysis of the filling stage in the injection molding process: Role of the mold-polymer interface, J. Appl. Polym. Sci., 121 (2011), 1579-1592. |
| [8] | A. Khalifeh, J. R. Clermont, Numerical simulations of non-isothermal three-dimensional flows in an extruder by a finite-volume method, J. Non-Newton. Fluid Mech., 126 (2005), 7-22. |
| [9] | P. S. B. Zdanski, M. Vaz Jr, Three-dimensional polymer melt flow in sudden expansions: Nonisothermal flow topology, Int. J. Heat Mass Tran., 52 (2009), 3585-3594. |
| [10] | A. K. Doufas, A. J. McHugh, C. Miller, Simulation of melt spinning including flow-induced crystallization Part Ⅰ. Model development and predictions, J. Non-Newton. Fluid Mech., 92 (2000), 27-66. |
| [11] | G. W. M. Peters, F. P. T. Baaijens, Modeling of non-isothermal viscoelastic flows, J. Non-Newton. Fluid Mech., 68 (1997), 205-224. |
| [12] | K. Kunisch, X. Marduel, Optimal control of non-isothermal viscoelastic fluid flow, J. Non-Newton. Fluid Mech., 88 (2000), 261-301. |
| [13] | A. Wachs, J. R. Clermont, Non-isothermal viscoelastic flow computations in an axisymmetric contraction at high Weissenberg numbers by a finite volume method, J. Non-Newton. Fluid Mech., 95 (2000), 147-184. |
| [14] | M. Laso, H. C. Öttinger, Calculation of viscoelastic flow using molecular models: the CONNFFESSIT approach, J. Non-Newton. Fluid Mech., 47 (1993), 1-20. |
| [15] | R. Keunings, Micro-macro methods for the multiscale simulation of viscoelastic flow using molecular models of kinetic theory, Rheology Reviews, 2004 (2004), 67-98. |
| [16] | M. A. Hulsen, A. P. G. van Heel, B. H. A. A. van den Brule, Simulation of viscoelastic flows using Brownian configuration fields, J. Non-Newton. Fluid Mech., 70 (1997), 79-101. |
| [17] | P. Halin, G. Lielens, R. Keunings, V. Legat, The Lagrangian particle method for macroscopic and micro-macro viscoelastic flow computations, J. Non-Newton. Fluid Mech., 79 (1998), 387-403. |
| [18] | W. E, B. Engquist, The heterogeneous multiscale methods, Commun. Math. Sci., 1 (2003), 87-132. |
| [19] | S. Yasuda, R. Yamamoto, A model for hybrid simulations of molecular dynamics and computational fluid dynamics, Phys. Fluids, 20 (2008), 113101. |
| [20] | T. Murashima, T. Taniguchi, Multiscale Lagrangian fluid dynamics simulation for polymeric fluid, J. Polym. Sci. B Polym. Phys., 48 (2010), 886-893. |
| [21] | T. Murashima, T. Taniguchi, Multiscale simulation of history-dependent flow in entangled polymer melts, Europhys. Lett., 96 (2011), 18002. |
| [22] | T. Murashima, T. Taniguchi, Flow-history-dependent behavior of entangled polymer melt flow analyzed by multiscale simulation, J. Phys. Soc. Jpn., 81 (2012), 1-7. |
| [23] | T. Sato, K. Harada, T. Taniguchi, Multiscale simulations of flows of a well-entangled polymer melt in a contraction-expansion channel, Macromolecules, 52 (2019), 547-564. |
| [24] | T. Sato, T. Taniguchi, Multiscale simulations for entangled polymer melt spinning process, J. NonNewton. Fluid Mech., 241 (2017), 34-42. |
| [25] | M. Doi, J. Takimoto, Molecular modeling of entanglement, Phil. Trans. R. Soc. Lond. A, 361 (2003), 641-652. |
| [26] | K. Kremer, G. S. Grest, Dynamics of entangled linear polymer melts: A molecular-dynamics simulation, J. Chem. Phys., 92 (1990), 5057-5086. |
| [27] | S. Yasuda, R. Yamamoto, Synchronized molecular-dynamics simulation via macroscopic heat and momentum transfer: An application to polymer lubrication, Phys. Rev. X, 4 (2014), 041011. |
| [28] | S. Yasuda, R. Yamamoto, Synchronized molecular-dynamics simulation for the thermal lubrication of a polymeric liquid between parallel plates, Comput. Fluids, 124 (2016), 185-189. |
| [29] | S. Yasuda, Synchronized molecular-dynamics simulation of the thermal lubrication of an entangled polymeric liquid, Polymers, 11 (2019), 131. |
| [30] | T. Sato, T. Taniguchi, Rheology and entanglement structure of well-entangled polymer melts: A slip-link simulation study, Macromolecules, 52 (2019), 3951-3964. |
| [31] | F. Habla, A. Woitalka, S. Neuner, O. Hinrichsen, Development of a methodology for numerical simulation of non-isothermal viscoelastic fluid flows with application to axisymmetric 4:1 contraction flows, Chem. Eng. J., 207-208 (2012), 772-784. |
| [32] | H. Pol, S. Banik, B. L. Azad, S. Thete, P. Doshi, A. Lele, Nonisothermal analysis of extrusion film casting process using molecular constitutive equations, Rheol. Acta, 53 (2014), 85-101. |
| [33] | M. Zatloukal, Measurements and modeling of temperature-strain rate dependent uniaxial and planar extensional viscosities for branched LDPE polymer melt, Polymer, 104 (2016), 258-267. |
| [34] | X. Zhuang, J. Ouyang, W. M. Li, Y. G. Li, Three-dimensional simulations of non-isothermal transient flow and flow-induced stresses during the viscoelastic fluid filling process, Int. J. Heat Mass Tran., 104 (2017), 374-391. |
| [35] | P. Gao, X. Wang, J. Ouyang, Numerical investigation of non-isothermal viscoelastic filling process by a coupled finite element and discontinuous Galerkin method, Int. J. Heat Mass Tran., 140 (2019), 227-242. |
| [36] | M. Doi, S. Edwards, The theory of polymer dynamics, Oxford University Press, 1986. |
| [37] | G. Marrucci, Dynamics of entanglements: A nonlinear model consistent with the Cox-Merz rule, J. Non-Newton. Fluid Mech., 62 (1996), 279-289. |
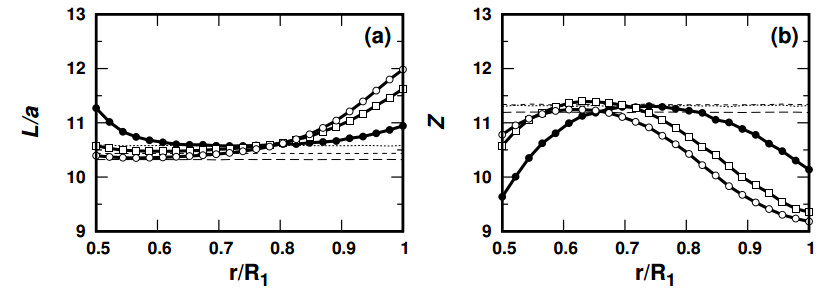
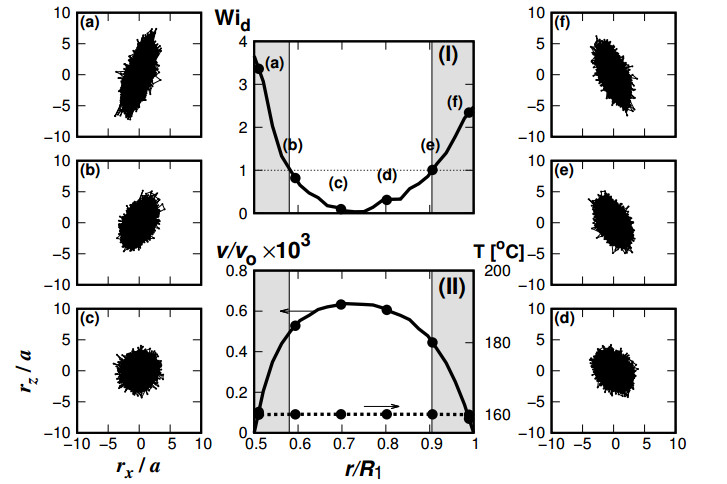
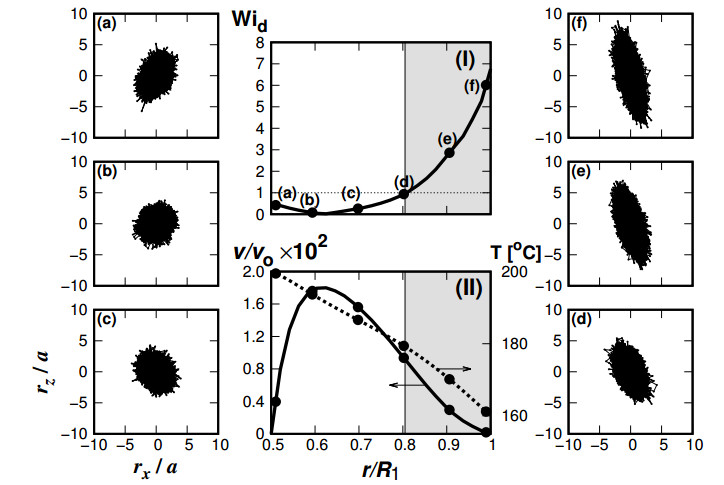
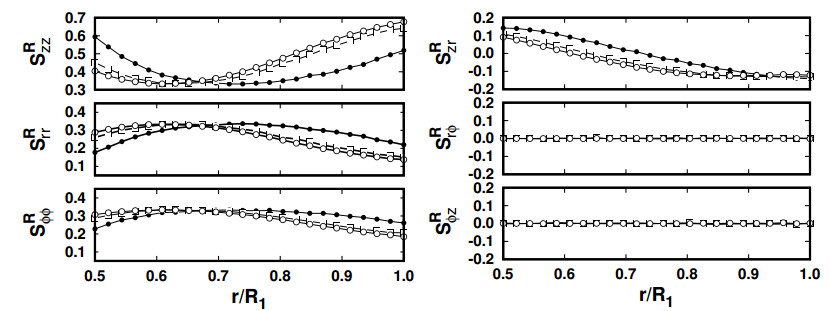
Figures(12)

Yuji Hamada, Takeshi Sato, Takashi Taniguchi. Multiscale simulation of a polymer melt flow between two coaxial cylinders under nonisothermal conditions[J]. Mathematics in Engineering, 2021, 3(6): 1-22. doi: 10.3934/mine.2021042







 DownLoad:
DownLoad: