Citation: Mostafa Adimy, Abdennasser Chekroun, Claudia Pio Ferreira. Global dynamics of a differential-difference system: a case of Kermack-McKendrick SIR model with age-structured protection phase[J]. Mathematical Biosciences and Engineering, 2020, 17(2): 1329-1354. doi: 10.3934/mbe.2020067

| [1] | N. T. J. Bailey, The mathematical theory of infectious diseases and its applications, Charles Griffin & Company Ltd, 1975. |
| [2] | H. W. Hethcote, The Mathematics of Infectious Diseases, SIAM Rev., 42 (2000), 599-653. |
| [3] | R. M. Anderson and R. M. May, Age-related changes in the rate of disease transmission: implications for the design of vaccination programmes, Epidemiol. Infect., 94 (1985), 365-436. |
| [4] | D. Schenzle, An Age-Structured Model of Pre- and Post-Vaccination Measles Transmission, Math. Med. Biol. J. IMA, 1 (1984), 169-191. |
| [5] | L. J. Allen and P. van den Driessche, The basic reproduction number in some discrete-time epidemic models, J. Differ. Equations Appl., 14 (2008), 1127-1147. |
| [6] | B. F. Finkenstadt and B. T. Grenfell, Time series modelling of childhood diseases: a dynamical systems approach, Appl. Statist., 49 (2000), 187-205. |
| [7] | R. M. Anderson and R. M. May, Population biology of infectious diseases: Part I, Nature, 280 (1979), 361-367. |
| [8] | F. Brauer and C. Castillo-Chavez, Mathematical models in population biology and epidemiology, 2nd edition, Texts in Applied Mathematics, Springer, New York, 2012. |
| [9] | W. O. Kermack and A. G. McKendrick, A contribuition to the mathematical theory of epidemics, Proc. R. Soc. Lond. A, 115 (1927), 700-721. |
| [10] | M. Martcheva, An Introduction to Mathematical Epidemiology, 1st edition, Springer Publishing Company, Incorporated, 2015. |
| [11] | J. Cushing, An Introduction to Structured Population Dynamics, SIAM, 1998. |
| [12] | O. Diekmann, J. A. P. Heesterbeek and J. A. J. Metz, On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations, J. Math. Biol., 28 (1990), 365-382. |
| [13] | H. R Thieme and C. Castillo-Chavez, How may infection-age-dependent infectivity affect the dynamics of HIV/AIDS?, SIAM J. Appl. Math., 53 (1993), 1447-1479. |
| [14] | H. R. Thieme, Mathematics in Population Biology, Princeton University Press, 2003. |
| [15] | E. Beretta and Y. Takeuchi, Global stability of an SIR epidemic model with time delays, J. Math. Biol., 33 (1995), 250-260. |
| [16] | V. Capasso and G. Serio, A generalization of the Kermack-McKendrick deterministic epidemic model, Math. Biosci., 42 (1978), 43-61. |
| [17] | K. L. Cooke and P. van den Driessche, Analysis of an SEIRS epidemic model with two delays, J. Math. Biol., 35 (1996), 240-260. |
| [18] | S. Ruan, D. Xiao and J. C. Beier, On the delayed Ross-Macdonald model for malaria transmission, Bull. Math. Biol., 70 (2008), 1098-1114. |
| [19] | L. J. Allen, An Introduction to Stochastic Epidemic Models, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, 81-130. |
| [20] | Y. Lin, D. Jiang and S. Wang, Stationary distribution of a stochastic SIS epidemic model with vaccination, Phys. A (Amsterdam, Neth.), 394 (2014), 187-197. |
| [21] | Y. Zhao and D. Jiang, The threshold of a stochastic SIS epidemic model with vaccination, Appl. Math. Comput., 243 (2014), 718-727. |
| [22] | R. M. Anderson and R. M. May, Spatial, temporal, and genetic heterogeneity in host populations and the design of immunization programs, IMA J. Math. Appl. Med. Biol., 1 (1994), 233-266. |
| [23] | A. Scherer and A. McLean, Mathematical models of vaccination, Br. Med. Bull., 62 (2002), 187-199. |
| [24] | L. Gao and H. Hethcote, Simulations of rubella vaccination strategies in China, Math. Biosci., 202 (2006), 371-385. |
| [25] | H. W. Hethcote, Optimal ages of vaccination for measles, Math. Biosci., 89 (1988), 29-52. |
| [26] | R. Donken, T. M. S. Klooster, R. M. Schepp, et al., Immune Responses After 2 Versus 3 Doses of HPV Vaccination up to 4 1/2 Years After Vaccination: An Observational Study Among Dutch Routinely Vaccinated Girls, J. Infect. Dis., 215 (2017), 359-367. |
| [27] | M. Stanley, HPV-immune response to infection and vaccination, Infect. Agents Cancer, 5 (2010), 19-25. |
| [28] | A. N. Burchell, R. L. Winer, S. de Sanjosé, et al., Epidemiology and transmission dynamics of genital HPV infection, Vaccine, 24 (2006), S52-S61. |
| [29] | P. L. Ho, E. N. Miyaji, M. L. S. Oliveira, et al., Economical Value of Vaccines for the Developing Countries-The Case of Instituto Butantan, a Public Institution in Brazil, PLoS Neglected Trop. Dis., 5 (2011), e1300. |
| [30] | L. Kubin, Is There a Resurgence of Vaccine Preventable Diseases in the US?, J. Pediatr. Nurs., 44 (2019), 115-118. |
| [31] | C. I. Paules, H. D. Marston and A. S. Fauci, Measles in 2019 - going backward, N. Engl. J. Med., 380 (2019), 2185-2187. |
| [32] |
R. Peralta, C. Vargas-De-León and P. Miramontes, Global Stability Results in a SVIR Epidemic Model with Immunity Loss Rat Depending on the Vaccine-Age, Abstract and Applied Analysis, 2015 (2015), 1-8. Available from: https://www.hindawi.com/journals/aaa/2015/341854/abs/. doi: 10.1155/2015/341854

|
| [33] | S. M. Raimundo, H. M. Yang and A. B. Engel, Modelling the effects of temporary immune protection and vaccination against infectious diseases, Appl. Math. Comput., 189 (2007), 1723-1736. |
| [34] | D. Bernoulli, Essai d'une nouvelle analyse de la mortalité causée par la petite vérole et des avantages de l'inoculation pour la prévenir, Histoire de l'Acad., Roy. Sci.(Paris) avec Mem, 1 (1760), 1-45. |
| [35] | X. Duan, S. Yuan and X. Li, Global stability of an SVIR model with age of vaccination, Appl. Math. Comput., 226 (2014), 528-540. |
| [36] | M. Iannelli, M. Martcheva and X. Z. Li, Strain replacement in an epidemic model with superinfection and perfect vaccination, Math. Biosci., 195 (2005), 23-46. |
| [37] | X. Z. Li, J. Wang and M. Ghosh, Stability and bifurcation of an SIVS epidemic model with treatment and age of vaccination, Appl. Math. Modell., 34 (2010), 437-450. |
| [38] | Q. Liu, D. Jiang, T. Hayat, et al., Analysis of a delayed vaccinated SIR epidemic model with temporary immunity and lévy jumps, Nonlinear Anal. Hybrid Syst., 27 (2018), 29-43. |
| [39] | J. Xu and Y. Zhou, Global stability of a multi-group model with generalized nonlinear incidence and vaccination age, Discrete Contin. Dyn. Sys. B, 21 (2016), 977-996. |
| [40] | G. Webb, Theory of Nonlinear Age-Dependent Population Dynamics, CRC Press, New York, 1985. |
| [41] | J. K. Hale and M. A. Cruz, Existence, uniqueness and continuous dependence for hereditary systems, Ann. Mat. Pura Appl., 85 (1970), 63-81. |
| [42] | J. K. Hale and S. M. Verduyn Lunel, Introduction to Functional Differential Equations, Springer, 1993. |
| [43] | H. Smith, An Introduction to Delay Differential Equations with Applications to the Life Sciences, Texts in Applied Mathematics, Springer New York, 2011. |
| [44] | M. Adimy, A. Chekroun and T. M. Touaoula, Age-structured and delay differential-difference model of hematopoietic stem cell dynamics, Discrete Contin. Dyn. Syst. Ser. B, 20 (2015), 2765-2791. |
| [45] | M. A. Cruz and J. K. Hale, Stability of functional differential equations of neutral type, J. Differ. Equations, 7 (1970), 334-355. |
| [46] | A. Perasso, An introduction to the basic reproduction number in mathematical epidemiology, ESAIM Proc. Surv., 62 (2018), 123-138. |
| [47] | H. I. Freedman and P. Moson, Persistence definitions and their connections, Proc. Am. Math. Soc., 109 (1990), 1025-1033. |
| [48] | K. Gu and Y. Liu, Lyapunov-Krasovskii functional for uniform stability of coupled differentialfunctional equations, Automatica, 45 (2009), 798-804. |
| [49] | M. Hou, G. Duan and M. Guo, New versions of Barbalat's lemma with applications, J. Control Theory Appl., 8 (2010), 545-547. |
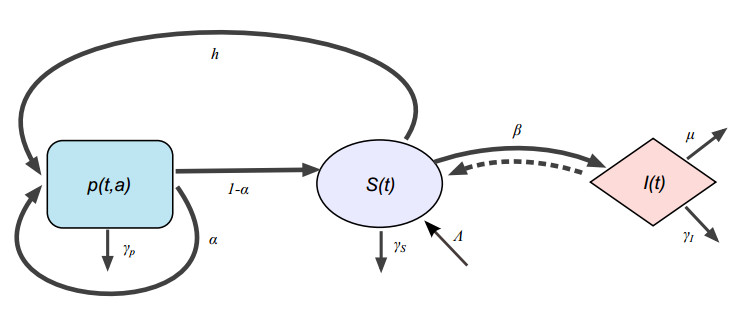
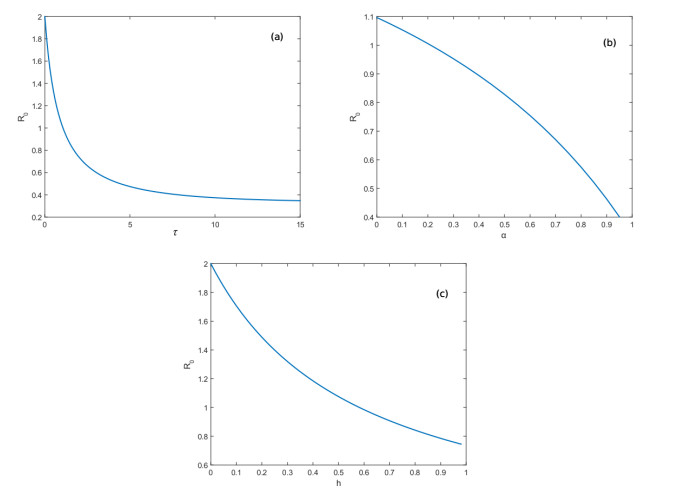
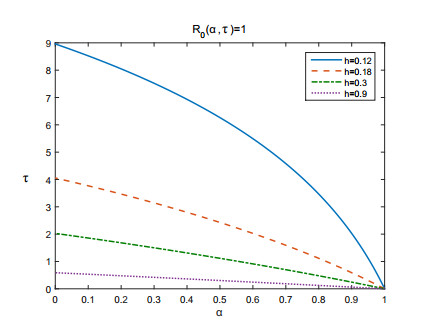
Figures(3) / Tables(1)

Mostafa Adimy, Abdennasser Chekroun, Claudia Pio Ferreira. Global dynamics of a differential-difference system: a case of Kermack-McKendrick SIR model with age-structured protection phase[J]. Mathematical Biosciences and Engineering, 2020, 17(2): 1329-1354. doi: 10.3934/mbe.2020067







 DownLoad:
DownLoad: