Citation: Qian Li, Yanni Xiao. Analysis of a mathematical model with nonlinear susceptibles-guided interventions[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5551-5583. doi: 10.3934/mbe.2019276

| [1] | R. M. Anderson and R. M. May, Infectious Diseases of Humans, Dynamics and Control, Oxford University, Oxford,1991. |
| [2] | V. Capasso, Mathematical Structures of Epidemic Systems, Lecture Notes in Biomathematics, vol. 97, Springer-Verlag, Berlin, 1993. |
| [3] | H. W. Hethcote, The mathematics of infectious diseases, SIAM Rev., 42 (2000). |
| [4] | O. Diekmann and J. A. P. Heesterbeek, Mathematical Epidemiology of Infectious Diseases. Model Building, Analysis and Interpretation, Wiley Ser. Math. Comput. Biol., John Wiley and Sons, Chichester, England, 2000. |
| [5] | F. Brauer and P. van den Driessche, Models for the transmission of disease with immigration of infectives, Math. Biosci., 171 (2001), 143–154. |
| [6] | M. A. Nowak and A. R. McLean, A mathematical model of vaccination against HIV to prevent the development of AIDS, Proc. Biol. Sci., 246 (1991), 141–146. |
| [7] | N. M. Ferguson, D. A. T. Cummings, S. Cauchemez, et al., Strategies for containing an emerging in fuenza pandemic in Southeast Asia, Nature, 437 (2005), 209–214. |
| [8] | Y. N. Xiao, T. T. Zhao and S. Y. Tang, Dynamics of an infectious disease with media/ psychology induced non-sooth incidence, Math. Biosci. Eng., 10 (2013), 445–461. |
| [9] | A. L. Wang and Y. N. Xiao, A Filippov system describing media effects on the spread of infectious diseases, Nonlinear Anal. Hybri., 11 (2014), 84–97. |
| [10] | J. A. Cui, X. X. Mu and H. Wan, Saturation recovery leads to multiple endemic equilibria and backward bifurcation, J. Theor. Biol., 254 (2008), 275–283. |
| [11] | J. L. Wang, S. Q. Liu, B. W. Zheng, et al., Qualitative and bifurcation analysis using an SIR model with a saturated treatment function, Math. Comput. Model., 55 (2012), 710–722. |
| [12] | X. Zhang and X. N. Liu, Backward bifurcation of an epidemic model with saturated treatment function, J. Math. Anal. Appl., 348 (2008), 433–443. |
| [13] | L. H. Zhou and M. Fan, Dynamics of an SIR epidemic model with limited medical resources revisited, Nonlinear Anal. Real., 13 (2012), 312–324. |
| [14] | H. Wan and J. A. Cui, Rich dynamics of an epidemic model with saturation recovery, J. Appl. Math., 314958 (2013). |
| [15] | Z. H. Zhang and Y. H. Suo, Qualitative analysis of a SIR epidemic model with saturated treatment rate, J. Appl. Math. Comput., 34 (2010), 177–194. |
| [16] | S. Y. Tang and R. A. Cheke, Stage-dependent impulsive models of integrated pest management (IPM) strategies and their dynamic consequences, J. Math. Biol., 50 (2005), 257–292. |
| [17] | S. Y. Tang, Y. N. Xiao and R. A. Cheke, Dynamical analysis of plant disease models with cultural control strategies and economic thresholds, Math. Comput. Simul., 80 (2010), 894–921. |
| [18] | A. B. Sabin, Measles, killer of millions in developing countries: strategies of elimination and continuing control, Eur. J. Epidemiol., 7 (1991), 1–22. |
| [19] | B. Shulgin, L. Stone and Z. Agur, Pulse vaccination strategy in the SIR epidemic model, Bull. Math. Biol., 60 (1998), 1123–1148. |
| [20] | L. Stone, B. Shulgin and Z. Agur, Theoretical examination of the pulse vaccination policy in the SIR epidemic model, Math. Comput. Model., 31 (2000), 207–215. |
| [21] | A. D'Onofrio, On pulse vaccination strategy in the SIR epidemic model with vertical transmission, Appl. Math. Lett., 18 (2005), 729–732. |
| [22] | F. L. Black, Measles endemicity in insular populations: Critical community size and its evolutionary implication, J. Theor. Biol., 11 (1966), 207–211. |
| [23] | W. J. Moss and P. Strebel, Biological feasibility of measles eradication, J. Infect. Dis., 204 (2011), 47–53. |
| [24] | Q. Q. Zhang, B. Tang and S. Y. Tang, Vaccination threshold size and backward bifurcation of SIR model with state-dependent pulse control, J. Theor. Biol., 455 (2018), 75–85. |
| [25] | Q. Li and Y. N. Xiao, Dynamical behaviour and bifurcation analysis of the SIR model with continuous treatment and state-dependent impulsive control, Int. J. Bifurcat. Chaos, (2019), Accepted. |
| [26] | A. L. Wang, Y. N. Xiao and R. Smith?, Using non-smooth models to determine thresholds for microbial pest management, J. Math. Biol., (2019). |
| [27] | A. L. Wang, Y. N. Xiao and R. Smith?, Multiple equilibria in a non-smooth epidemic model with medical-resource constraints, Bull. Math. Biol., 81 (2019), 963–994. |
| [28] | W. J. Qin, S. Y. Tang and R. A. Cheke, Nonlinear pulse vaccination in an SIR epidemic model with resource limitation, Abstr. Appl. Anal., 670263 (2013). |
| [29] | J. Yang and S. Y. Tang, Holling type II predator–prey model with nonlinear pulse as state-dependent feedback control, J. Comput. Appl. Math., 291 (2016), 225–241. |
| [30] | S. Y. Tang, B. Tang, A. L. Wang, et al., Holling II predator-prey impulsive semi-dynamic model with complex Poincaré map, Nonlinear Dyn., 81 (2015), 1575–1596. |
| [31] | S. Y. Tang, W. H. Pang, R. A. Cheke, et al., Global dynamics of a state-dependent feedback control system, Adv. Diff. Equ., 322 (2015), DOI: 10.1186/s13662-015-0661-x. |
| [32] | J. M. Grandmont, Nonlinear difference equations, bifurcations and chaos: an introduction, Research in Economics, 62 (2008), 122–177. |
| [33] | Y. N. Xiao, X. X. Xu and S. Y. Tang, Sliding mode control of outbreaks of emerging infectious diseases, Bull. Math. Biol., 74 (2012), 2403–2422. |
| [34] | Y. P. Yang, Y. N. Xiao and J. H. Wu, Pulse HIV vaccination: feasibility for virus eradication and optimal vaccination schedule, Bull. Math. Biol., 75 (2013), 725–751. |
| [35] | Y. Tian, K. B. Sun, A. Kasperski, et al., Nonlinear modelling and qualitative analysis of a real chemostat with pulse feeding, Discrete Dyn. Nat. Soc., 640594 (2011), 1–18. |
| [36] | S. Y. Tang and W. H. Pang, On the continuity of the function describing the times of meeting impulsive set and its application, Math. Biosci. Eng., 14 (2017), 1399–1406. |
| [37] | X. N. Liu, Y. Takeuchi and S. Iwami, SVIR epidemic model with vaccination strategy, J. Theor. Biol., 253 (2008), 1–11. |

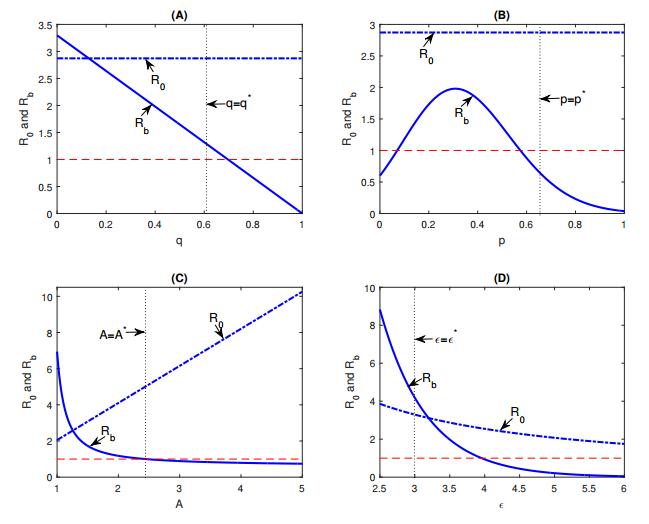
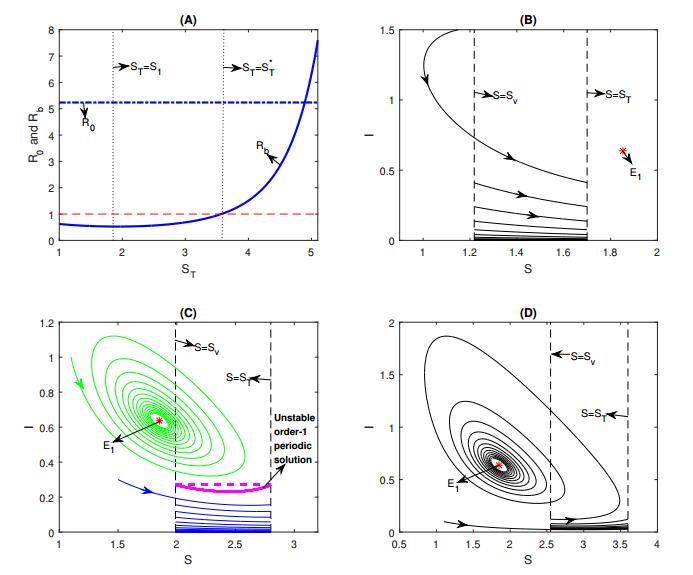
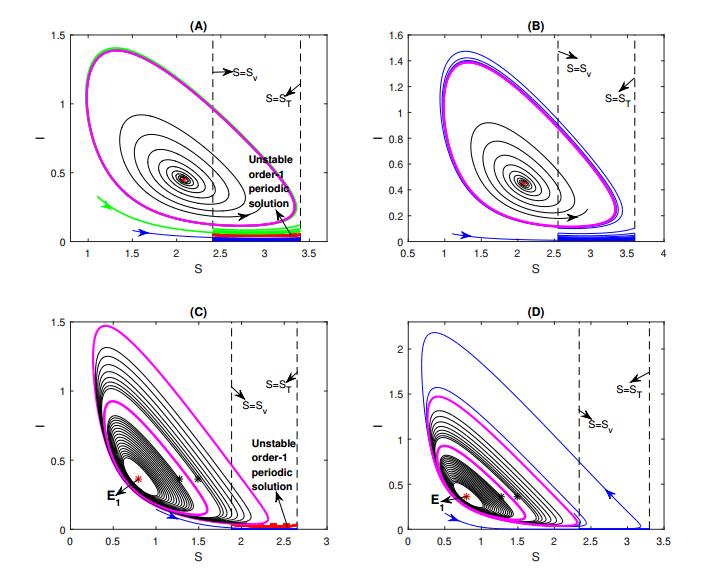

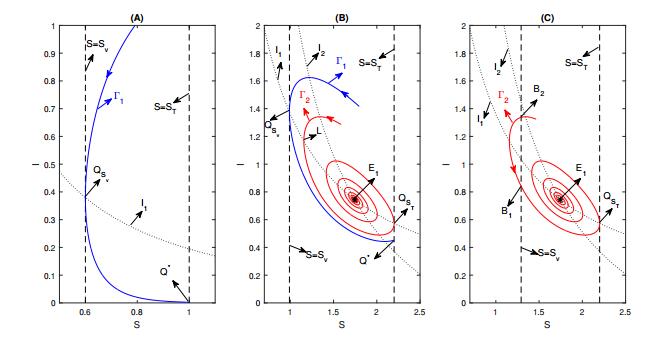
Figures(8) / Tables(1)

Qian Li, Yanni Xiao. Analysis of a mathematical model with nonlinear susceptibles-guided interventions[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5551-5583. doi: 10.3934/mbe.2019276







 DownLoad:
DownLoad: