Citation: Krzysztof Fujarewicz. Estimation of initial functions for systems with delays from discrete measurements[J]. Mathematical Biosciences and Engineering, 2017, 14(1): 165-178. doi: 10.3934/mbe.2017011

| [1] | [ M. Anguelova,B. Wennberg, State elimination and identifiability of the delay parameter for nonlinear time-delay systems, Automatica, 44 (2008): 1373-1378. |
| [2] | [ C. T. H. Baker,E. I. Parmuzin, Identification of the initial function for nonlinear delay differential equations, Russ. J. Numer. Anal. Math. Modelling, 20 (2005): 45-66. |
| [3] | [ C. T. H. Baker,E. I. Parmuzin, Initial function estimation for scalar neutral delay differential equations, Russ. J. Numer. Anal. Math. Modelling, 23 (2008): 163-183. |
| [4] | [ L. Belkoura,J. P. Richard,M. Fliess, Parameters estimation of systems with delayed and structured entries, Automatica, 45 (2009): 1117-1125. |
| [5] | [ K. Fujarewicz,A. Galuszka, Generalized backpropagation through time for continuous time neural networks and discrete time measurements, Artificial Intelligence and Soft Computing -ICAISC 2004 (eds. L. Rutkowski, J. Siekmann, R. Tadeusiewicz and L. A. Zadeh), Lecture Notes in Computer Science, 3070 (2004): 190-196. |
| [6] | [ K. Fujarewicz,M. Kimmel,A. Swierniak, On fitting of mathematical models of cell signaling pathways using adjoint systems, Math. Biosci. Eng., 2 (2005): 527-534. |
| [7] | [ K. Fujarewicz,M. Kimmel,T. Lipniacki,A. Swierniak, Adjoint systems for models of cell signalling pathways and their application to parametr fitting, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 4 (2007): 322-335. |
| [8] | [ K. Fujarewicz,K. Lakomiec, Parameter estimation of systems with delays via structural sensitivity analysis, Discrete and Continuous Dynamical Systems -series B, 19 (2014): 2521-2533. |
| [9] | [ K. Fujarewicz,K. Lakomiec, Adjoint sensitivity analysis of a tumor growth model and its application to spatiotemporal radiotherapy optimization, Mathematical Biosciences and Engineering, 13 (2016): 1131-1142. |
| [10] | [ M. Jakubczak,K. Fujarewicz, Application of adjoint sensitivity analysis to parameter estimation of age-structured model of cell cycle, in Information Technologies in Medicine, (eds. E. Pietka, P. Badura, J. Kawa and W. Wieclawek), Advances in Intelligent Systems and Computing, 472 (2016): 123-131. |
| [11] | [ K. Ł akomiec, S. Kumala, R. Hancock, J. Rzeszowska-Wolny and K. Fujarewicz, Modeling the repair of DNA strand breaks caused by $γ$-radiation in a minichromosome, Physical Biology 11 (2014), 045003. |
| [12] | [ M. Liu,Q. G. Wang,B. Huang,C. C. Hang, Improved identification of continuous-time delay processes from piecewise step tests, Journal of Process Control, 17 (2007): 51-57. |
| [13] | [ R. Loxton,K. L. Teo,V. Rehbock, An optimization approach to state-delay identification, IEEE Transactions on Automatic Control, 55 (2010): 2113-2119. |
| [14] | [ B. Ni,D. Xiao,S. L. Shah, Time delay estimation for MIMO dynamical systems with time-frequency domain analysis, Journal of Process Control, 20 (2010): 83-94. |
| [15] | [ B. Rakshit,A. R. Chowdhury,P. Saha, Parameter estimation of a delay dynamical system using synchronization inpresence of noise, Chaos, Solitons and Fractals, 32 (2007): 1278-1284. |
| [16] | [ J. P. Richard, Time-delay systems: An overview of some recent advances and open problems, Automatica, 39 (2003): 1667-1694. |
| [17] | [ Y. Tang,X. Guan, Parameter estimation of chaotic system with time-delay: A differential evolution approach, Chaos, Solitons and Fractals, 42 (2009): 3132-3139. |
| [18] | [ Y. Tang,X. Guan, Parameter estimation for time-delay chaotic systems by particle swarm optimization, Chaos, Solitons and Fractals, 40 (2009): 1391-1398. |
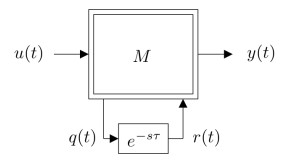
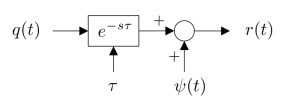

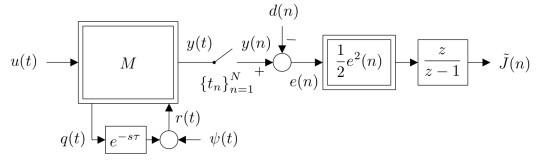
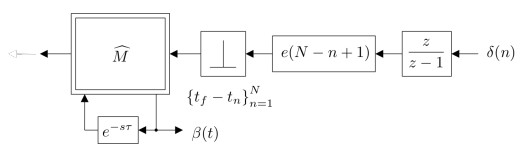
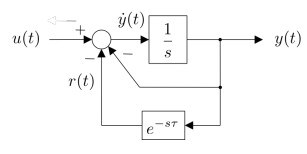
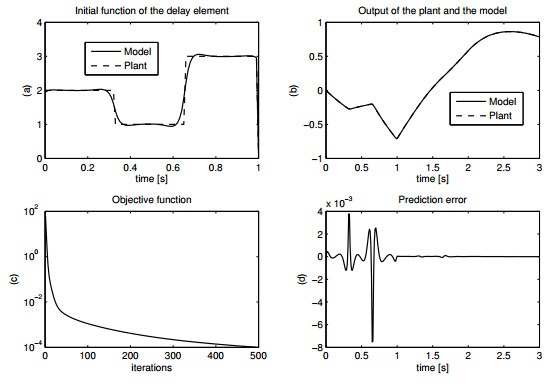
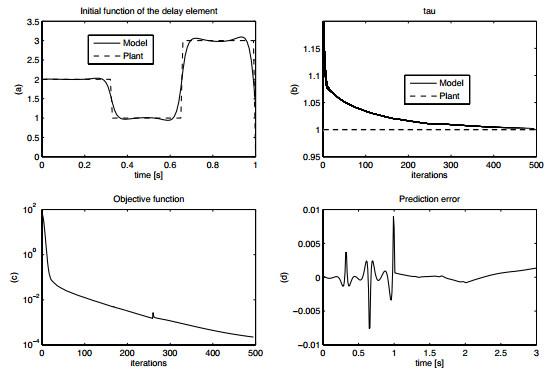
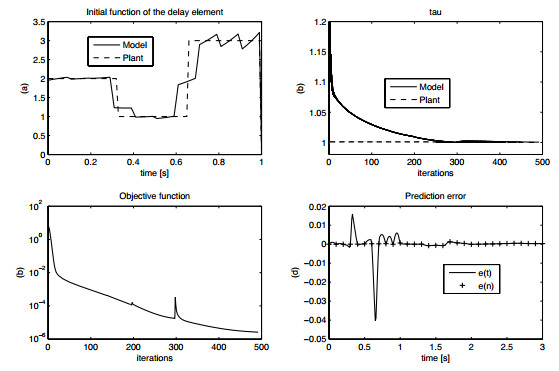
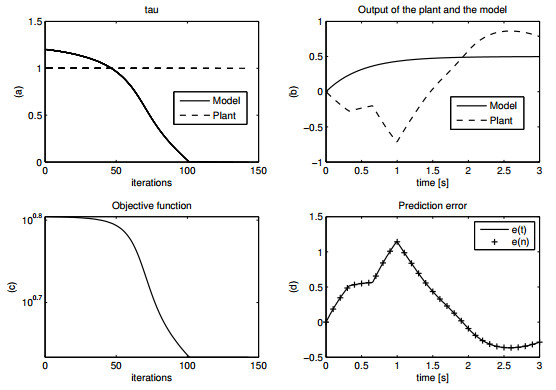
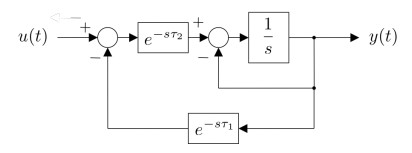
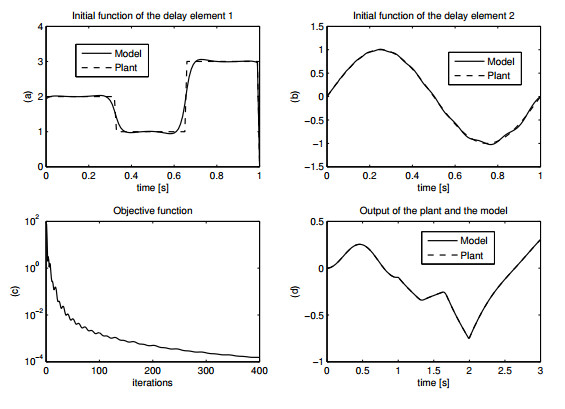
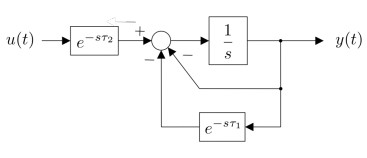
Figures(15) / Tables(1)

Krzysztof Fujarewicz. Estimation of initial functions for systems with delays from discrete measurements[J]. Mathematical Biosciences and Engineering, 2017, 14(1): 165-178. doi: 10.3934/mbe.2017011







 DownLoad:
DownLoad: