Citation: Yongxing Li, Jianye Yang, Yufen Xu, Ming Zhang, Xiaoping Zhang, Wenyu Chen, Xiaodong Lv. A meta-analysis of the comparing of the first-generation and next-generation TKIs in the treatment of NSCLC[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5687-5696. doi: 10.3934/mbe.2019283

| [1] | T. J. Lynch, D. W. Bell, R. Sordella, et al., Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib, N. Engl. J. Med., 350 (2004), 2129–2139. |
| [2] | Y. L. Wu, W. Z. Zhong, L. Y. Li, et al., Epidermal growth factor receptor mutations and their correlation with gefitinib therapy in patients with non-small cell lung cancer: a meta-analysis based on updated individual patient data from six medical centers in mainland China, J. Thorac. Oncol., 2 (2007), 430–439. |
| [3] | R. Rosell, E. Carcereny, R. Gervais, et al., Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial, Lancet Oncol., 13 (2012), 239–246. |
| [4] | T. S. Mok, Y. L. Wu, S. Thongprasert, et al., Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma, N. Engl. J. Med., 361 (2009), 947–957. |
| [5] | L. V. Sequist, J. C. Yang, N. Yamamoto, et al., Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations, J. Clin. Oncol., 31 (2013), 3327–3334. |
| [6] | G. Recondo, F. Facchinetti, K. A. Olaussen, et al., Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI?, Nat. Rev. Clin. Oncol., 15 (2018), 694–708. |
| [7] | K. Park, E. H. Tan, K. O'Byrne, et al., Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial, Lancet Oncol., 17 (2016), 577–589. |
| [8] | L. Paz-Ares, E. H. Tan, K. O'Byrne, et al., Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial, Ann. Oncol., 28 (2017), 270–277. |
| [9] | A. R. Jadad, R. A. Moore, D. Carroll, et al., Assessing the quality of reports of randomized clinical trials: is blinding necessary?, Control. Clin. Trials, 17 (1996), 1–12. |
| [10] | J. P. Higgins and S. G. Thompson, Quantifying heterogeneity in a meta-analysis, Stat. Med., 21 (2002), 1539–1558. |
| [11] | J. P. Higgins, S. G. Thompson, J. J. Deeks, et al., Measuring inconsistency in meta-analyses, BMJ, 327 (2003), 557–560. |
| [12] | J. C. Soria, Y. Ohe, J. Vansteenkiste, et al., Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer, N. Engl. J. Med., 378 (2018), 113–125. |
| [13] | T. S. Mok, Y. Cheng, X. Zhou, et al., Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations, J. Clin. Oncol., 36 (2018), 2244–2250. |
| [14] | Y. L. Wu, Y. Cheng, X. Zhou, et al., Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial, Lancet Oncol., 18 (2017), 1454–1466. |
| [15] | S. Novello, F. Barlesi, R. Califano, et al., Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up, Ann. Oncol., 27 (2016), v1–v27. |
| [16] | H. A. Yu, M. E. Arcila, N. Rekhtman, et al., Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers, Clin. Cancer Res., 19 (2013), 2240–2247. |
| [17] | S. N. Kazaz and I. Oztop, Treatment after first-generation epidermal growth factor receptor tyrosine kinase inhibitor resistance in non-small-cell lung cancer, Turk. Thorac. J., 18 (2017), 66–71. |
| [18] | L. V. Sequist, B. A. Waltman, D. Dias-Santagata, et al., Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors, Sci. Transl. Med., 3 (2011), 75ra26. |
| [19] | C. H. Yun, K. E. Mengwasser, A. V. Toms, et al., The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP, Proc. Natl. Acad. Sci. USA, 105 (2008), 2070–2075. |
| [20] | A. Michalczyk, S. Kluter, H. B. Rode, et al., Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR, Bioorg. Med. Chem., 16 (2008), 3482–3488. |
| [21] | M. L. Sos, H. B. Rode, S. Heynck, et al., Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation, Cancer Res., 70 (2010), 868–874. |
| [22] | D. Li, L. Ambrogio, T. Shimamura, et al., BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models, Oncogene, 27 (2008), 4702–4711. |
| [23] | F. Solca, G. Dahl, A. Zoephel, et al., Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker, J. Pharmacol. Exp. Ther., 343 (2012), 342–350. |
| [24] | D. A. Cross, S. E. Ashton, S. Ghiorghiu, et al., AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discov., 4 (2014), 1046–1061. |
| [25] | Y. L. Wu, M. J. Ahn, M. C. Garassino, et al., CNS efficacy of osimertinib in patients with t790m-positive advanced non-small-cell lung cancer: data from a randomized phase III trial (AURA3), J. Clin. Oncol., 36 (2018), 2702–2709. |
| [26] | O. Romanidou, L. Landi, F. Cappuzzo, et al., Overcoming resistance to first/second generation epidermal growth factor receptor tyrosine kinase inhibitors and ALK inhibitors in oncogene-addicted advanced non-small cell lung cancer, Ther. Adv. Med. Oncol., 8 (2016), 176–187. |
| [27] | C. Zhou and L. D. Yao, Strategies to Improve Outcomes of Patients with EGRF-Mutant Non-Small Cell Lung Cancer: Review of the Literature, J. Thorac. Oncol., 11 (2016), 174–186. |
| [28] | H. Shigematsu, L. Lin, T. Takahashi, et al., Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers, J. Natl. Cancer Inst., 97 (2005), 339–346. |
| [29] | C. K. Lee, Y. L. Wu, P. N. Ding, et al., Impact of specific epidermal growth factor receptor (egfr) mutations and clinical characteristics on outcomes after treatment with egfr tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: A Meta-analysis, J. Clin. Oncol., 33 (2015), 1958–1965. |
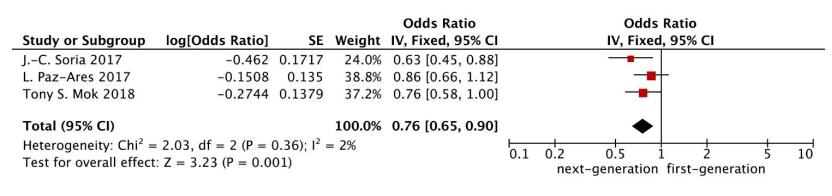
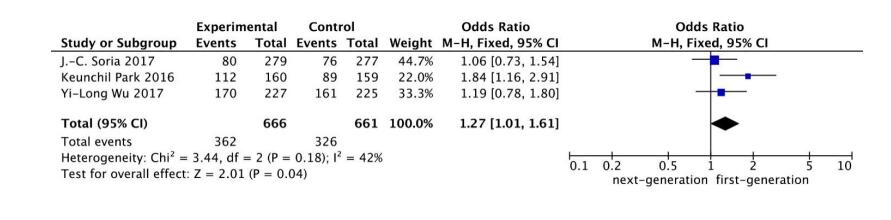
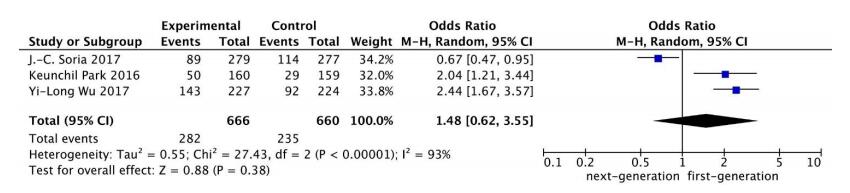
Figures(7) / Tables(1)

Yongxing Li, Jianye Yang, Yufen Xu, Ming Zhang, Xiaoping Zhang, Wenyu Chen, Xiaodong Lv. A meta-analysis of the comparing of the first-generation and next-generation TKIs in the treatment of NSCLC[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5687-5696. doi: 10.3934/mbe.2019283







 DownLoad:
DownLoad: