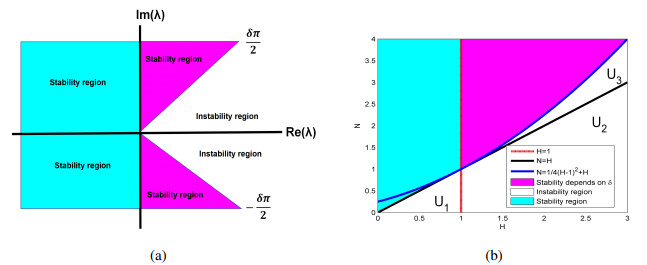
This paper contributes to the field by developing a fractional-order vegetation-sand model that incorporates memory effects into the traditional integer-order framework. By studying the spatiotemporal dynamics of a time-order fractional vegetation-sand model, the research aimed to deepen our understanding of the complex interactions between vegetation and sand environments, providing insights for effective management and conservation strategies in arid and semi-arid regions. First, using the linear stability theory of fractional differential equations, we conducted a stability analysis of the spatially homogeneous fractional-order vegetation-sand model and provided the parametric conditions for stability and instability. Next, we performed a stability analysis of the spatiotemporal model, utilizing Turing instability to reveal the effects of diffusion and fractional order on vegetation distribution. Through numerical simulations, we demonstrated the spatiotemporal evolution patterns of the model under different environmental conditions and discussed the implications of these dynamic changes for ecological restoration and land management.
Citation: Yimamu Maimaiti, Zunyou Lv, Ahmadjan Muhammadhaji, Wang Zhang. Analyzing vegetation pattern formation through a time-ordered fractional vegetation-sand model: A spatiotemporal dynamic approach[J]. Networks and Heterogeneous Media, 2024, 19(3): 1286-1308. doi: 10.3934/nhm.2024055
This paper contributes to the field by developing a fractional-order vegetation-sand model that incorporates memory effects into the traditional integer-order framework. By studying the spatiotemporal dynamics of a time-order fractional vegetation-sand model, the research aimed to deepen our understanding of the complex interactions between vegetation and sand environments, providing insights for effective management and conservation strategies in arid and semi-arid regions. First, using the linear stability theory of fractional differential equations, we conducted a stability analysis of the spatially homogeneous fractional-order vegetation-sand model and provided the parametric conditions for stability and instability. Next, we performed a stability analysis of the spatiotemporal model, utilizing Turing instability to reveal the effects of diffusion and fractional order on vegetation distribution. Through numerical simulations, we demonstrated the spatiotemporal evolution patterns of the model under different environmental conditions and discussed the implications of these dynamic changes for ecological restoration and land management.

| [1] |
M. M. Kling, D. D. Ackerly, Global wind patterns and the vulnerability of wind-dispersed species to climate change, Nat. Clim. Change, 10 (2020), 868–875. https://doi.org/10.1038/s41558-020-0848-3 doi: 10.1038/s41558-020-0848-3

|
| [2] |
J. J. Whicker, D. D. Breshears, P. T. Wasiolek, T. B. Kirchner, R. A. Tavani, D. A. Schoep, et al., Temporal and spatial variation of episodic wind erosion in unburned and burned semiarid shrubland, J. Environ. Qual., 31 (2002), 599–612. https://doi.org/10.2134/jeq2002.5990 doi: 10.2134/jeq2002.5990
|
| [3] |
V. Podsetchine, G. Schernewski, The influence of spatial wind inhomogeneity on flow patterns in a small lake, Water Res., 33 (1999), 3348–3356. https://doi.org/10.1016/S0043-1354(99)00035-4 doi: 10.1016/S0043-1354(99)00035-4
|
| [4] |
A. Miri, D. Dragovich, Z. Dong, Wind-borne sand mass flux in vegetated surfaces–wind tunnel experiments with live plants, Catena, 172 (2019), 421–434. https://doi.org/10.1016/j.catena.2018.09.006 doi: 10.1016/j.catena.2018.09.006
|
| [5] |
J. Gao, D. M. Kennedy, S. McSweeney, Patterns of vegetation expansion during dune stabilization at the decadal scale, Earth Surf. Processes Landforms, 48 (2023), 3059–3073. https://doi.org/10.1002/esp.5681 doi: 10.1002/esp.5681
|
| [6] |
C. A. Klausmeier, Regular and irregular patterns in semiarid vegetation, Science, 284 (1999), 1826–1828. https://doi.org/10.1126/science.284.5421.1826 doi: 10.1126/science.284.5421.1826
|
| [7] |
J. Von Hardenberg, E. Meron, M. Shachak, Y. Zarmi, Diversity of vegetation patterns and desertification, Phys. Rev. Lett., 87 (2001), 198101. https://doi.org/10.1103/PhysRevLett.87.198101 doi: 10.1103/PhysRevLett.87.198101
|
| [8] |
M. Rietkerk, M. C. Boerlijst, F. Van Langevelde, R. HilleRisLambers, J. de Koppel, L. Kumar, et al., Self-organization of vegetation in arid ecosystems, Am. Nat., 160 (2002), 524–530. https://doi.org/10.1086/342078 doi: 10.1086/342078
|
| [9] |
R. HilleRisLambers, M. Rietkerk, F. van den Bosch, H. H. T. Prins, H. de Kroon, Vegetation pattern formation in semi-arid grazing systems, Ecology, 82 (2001), 50–61. https://doi.org/10.1890/0012-9658(2001)082[0050:VPFISA]2.0.CO; 2 doi: 10.1890/0012-9658(2001)082[0050:VPFISA]2.0.CO;2
|
| [10] |
F. Zhang, H. Zhang, M. R. Evans, T. Huang, Vegetation patterns generated by a wind-driven sand-vegetation system in arid and semi-arid areas, Ecol. Complexity, 31 (2017), 21–33. https://doi.org/10.1016/j.ecocom.2017.02.005 doi: 10.1016/j.ecocom.2017.02.005
|
| [11] |
Y. Maimaiti, W. Yang, J. Wu, Turing instability and coexistence in an extended Klausmeier model with nonlocal grazing, Nonlinear Anal. Real World Appl., 64 (2022), 103443. https://doi.org/10.1016/j.nonrwa.2021.103443 doi: 10.1016/j.nonrwa.2021.103443
|
| [12] |
G. Guo, S. Zhao, J. Wang, Y. Gao, Positive steady-state solutions for a water-vegetation model with the infiltration feedback effect, Discrete Contin. Dyn. Syst. - Ser. B, 29 (2024), 426–458. https://doi.org/10.3934/dcdsb.2023101 doi: 10.3934/dcdsb.2023101
|
| [13] |
G. Guo, S. Zhao, D. Pang, Y. Su, Stability and cross-diffusion-driven instability for a water-vegetation model with the infiltration feedback effect, Z. Angew. Math. Phys., 75 (2024), 33. https://doi.org/10.1007/s00033-023-02167-7 doi: 10.1007/s00033-023-02167-7
|
| [14] |
Y. Maimaiti, W. Yang, Spatial vegetation pattern formation and transition of an extended water–plant model with nonlocal or local grazing, Nonlinear Dyn., 112 (2024), 5765–5791. https://doi.org/10.1007/s11071-024-09299-z doi: 10.1007/s11071-024-09299-z
|
| [15] |
C. R. Tian, Turing pattern formation in a semiarid vegetation model with fractional-in-space diffusion, Bull. Math. Biol., 77 (2015), 2072–2085. https://doi.org/10.1007/s11538-015-0116-2 doi: 10.1007/s11538-015-0116-2
|
| [16] |
F. Zhang, L. Yao, W. Zhou, Q. You, H. Zhang, Using Shannon entropy and contagion index to interpret pattern self-organization in a dynamic vegetation-sand model, IEEE Access, 8 (2020), 17221–17230. https://doi.org/10.1109/access.2020.2968242 doi: 10.1109/access.2020.2968242
|
| [17] |
F. Zhang, Y. Li, Y. Zhao, Z. Liu, Vegetation pattern formation and transition caused by cross-diffusion in a modified vegetation-sand model, Int. J. Bifurcation Chaos, 32 (2022), 2250069. https://doi.org/10.1142/S0218127422500699 doi: 10.1142/S0218127422500699
|
| [18] |
J. Li, G. Guo, H. Yuan, Nonlocal delay gives rise to vegetation patterns in a vegetation-sand model, Math. Biosci. Eng., 21 (2024), 4521–4553. https://doi.org/10.3934/mbe.2024200 doi: 10.3934/mbe.2024200
|
| [19] | D. Matignon, Stability results for fractional differential equations with applications to control processing, Comput. Eng. Syst. Appl., 2 (1996), 963–968. |
| [20] | I. Petráš, Fractional-Order Nonlinear Systems: Modeling, Analysis and Simulation, Springer Science and Business Media, 2011. https://doi.org/10.1007/978-3-642-18101-6 |
| [21] |
M. S. Abdelouahab, N. E. Hamri, J. Wang, Hopf bifurcation and chaos in fractional-order modified hybrid optical system, Nonlinear Dyn., 69 (2012), 275–284. https://doi.org/10.1007/s11071-011-0263-4 doi: 10.1007/s11071-011-0263-4
|
| [22] |
V. Gafiychuk, B. Datsko, Inhomogeneous oscillatory structures in fractional reaction–diffusion systems, Phys. Lett. A, 372 (2008), 619–622. https://doi.org/10.1016/j.physleta.2007.07.071 doi: 10.1016/j.physleta.2007.07.071
|
| [23] |
A. Alsaedi, B. Ahmad, M. Kirane, R. Lassoued, Global existence and large time behavior of solutions of a time fractional reaction diffusion system, Fract. Calc. Appl. Anal., 23 (2020), 390–407. https://doi.org/10.1515/fca-2020-0019 doi: 10.1515/fca-2020-0019
|
| [24] |
B. Liu, R. Wu, Ch. Li, Patterns induced by super cross-diffusion in a predator-prey system with Michaelis-Menten type harvesting, Math. Biosci., 298 (2018), 71–79. https://doi.org/10.1016/j.mbs.2018.02.002 doi: 10.1016/j.mbs.2018.02.002
|
| [25] |
C. L. Li, X. G. Tian, T. H. He, New insights on piezoelectric thermoelastic coupling and transient thermo-electromechanical responses of multi-layered piezoelectric laminated composite structure, Eur. J. Mech. A. Solids, 91 (2021), 104416. https://doi.org/10.1016/j.euromechsol.2021.104416 doi: 10.1016/j.euromechsol.2021.104416
|
| [26] |
B. Liu, R. Wu, L. Chen, Turing-Hopf bifurcation analysis in a superdiffusive predator-prey model, Chaos: Interdiscipl. J. Nonlinear Sci., 28 (2018), 113118. https://doi.org/10.1063/1.5055711 doi: 10.1063/1.5055711
|
| [27] |
X. L. Gao, H. L. Zhang, Y. L. Wang, Z. Y. Li, Research on pattern dynamics behavior of a fractional vegetation-water model in arid flat environment, Fractal Fract., 8 (2024), 264. https://doi.org/10.3390/fractalfract8050264 doi: 10.3390/fractalfract8050264
|
| [28] |
S. Djilali, B. Ghanbari, S. Bentout, A. Mezouaghi, Turing-Hopf bifurcation in a diffusive mussel-algae model with time-fractional-order derivative, Chaos, Solitons Fractals, 138 (2020), 109954. https://doi.org/10.1016/j.chaos.2020.109954 doi: 10.1016/j.chaos.2020.109954
|
| [29] |
M. Caputo, M. Fabrizio, A new definition of fractional derivative without singular kernel, Prog. Fract. Differ. Appl., 1 (2015), 73–85. https://doi.org/10.12785/pfda/010201 doi: 10.12785/pfda/010201
|
| [30] |
V. Gafiychuk, B. Datsko, Inhomogeneous oscillatory solutions in fractional reaction–diffusion systems and their computer modeling, Appl. Math. Comput., 198 (2008), 251–260. https://doi.org/10.1016/j.amc.2007.08.065 doi: 10.1016/j.amc.2007.08.065
|
| [31] |
V. Gafiychuk, B. Datsko, V. Meleshko, D. Blackmore, Analysis of the solutions of coupled nonlinear fractional reaction–diffusion equations, Chaos, Solitons Fractals, 41 (2009), 1095–1104. https://doi.org/10.1016/j.chaos.2008.04.039 doi: 10.1016/j.chaos.2008.04.039
|
| [32] |
C. L. Li, J. H. Liu, T. H. He, Fractional-order rate-dependent thermoelastic diffusion theory based on new definitions of fractional derivatives with non-singular kernels and the associated structural transient dynamic responses analysis of sandwich-like composite laminates, Commun. Nonlinear Sci. Numer. Simul., 132 (2024), 107896. https://doi.org/10.1016/j.cnsns.2024.107896 doi: 10.1016/j.cnsns.2024.107896
|
| [33] |
J. Zou, D. F. Luo, A new result on averaging principle for Caputo-type fractional delay stochastic differential equations with Brownian motion, Appl. Anal., 103 (2024), 1397–1417. https://doi.org/10.1080/00036811.2023.2245845 doi: 10.1080/00036811.2023.2245845
|
| [34] |
W. M. An, D. F. Lou, J. Z. Huang, Relative controllability and Hyers-Ulam stability of Riemann-Liouville fractional delay differential system, Qual. Theory Dyn. Syst., 23 (2024), 180. https://doi.org/10.1007/s12346-024-01046-4 doi: 10.1007/s12346-024-01046-4
|
| [35] |
S. O. Edeki, O. A. Grace, Local fractional operator for a one-dimensional coupled burger equation of non-integer time order parameter, J. Math. Fundam. Sci., 50 (2018), 28–39. https://doi.org/10.5614/j.math.fund.sci.2018.50.1.3 doi: 10.5614/j.math.fund.sci.2018.50.1.3
|






Figures(7)
Yimamu Maimaiti, Zunyou Lv, Ahmadjan Muhammadhaji, Wang Zhang. Analyzing vegetation pattern formation through a time-ordered fractional vegetation-sand model: A spatiotemporal dynamic approach[J]. Networks and Heterogeneous Media, 2024, 19(3): 1286-1308. doi: 10.3934/nhm.2024055







 DownLoad:
DownLoad: