Citation: Kaiyin Zhou, YuxingWang, Sheng Zhang, Mina Gachloo, Jin-Dong Kim, Qi Luo, Kevin Bretonnel Cohen, Jingbo Xia. GOF/LOF knowledge inference with tensor decomposition in support of high order link discovery for gene, mutation and disease[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1376-1391. doi: 10.3934/mbe.2019067

| [1] | M. Simsek, B. Meijer, A. A. van Bodegraven, N. K de Boer, and J. J. M. Chris, Finding hidden treasures in old drugs: the challenges and importance of licensing generics, Drug Discov. Today, 23(2018), 17–21. |
| [2] | N C. Baker, S. Ekins, A. J. Williams and A. Tropsha, A bibliometric review of drug repurposing, Drug Discov. Today, (2018). |
| [3] | F. L. Hitchcock, The expression of a tensor or a polyadic as a sum of products, J. Math. Phys., 6 (1927), 164–189. |
| [4] | N. Maximilian, V. Tresp and H. P. Kriegel, A three-Way model for collective learning on multirelational data, ICML, 11 (2011), 809–816. |
| [5] | N. Madhav, M. Gupta and P. Talukdar, Higher-order relation schema induction using tensor factorization with back-o and aggregation, In Proceedings of the 56th Annual Meeting of the Association for Computational Linguistics, 1 (2018), 1575–1584. |
| [6] | N. Madhav, U. S. Saini and P. Talukdar, Relation schema induction using tensor factorization with side information, arXiv preprint, arXiv:1605.04227 (2016). |
| [7] | L. Timothée, N. Usunier and G. Obozinski, Canonical tensor decomposition for knowledge base completion, arXiv preprint, arXiv:1806.07297 (2018). |
| [8] | J. C. Ho, J. Ghosh, S. R. Steinhubl,W. F. Stewart, J. C. Denny, B. A. Malin and J Sun, Limestone: High-throughput candidate phenotype generation via tensor factorization, J. Biomed. Inform., 52 (2014), 199–211. |
| [9] | J. Fang and W. Jonathan, Tightly integrated genomic and epigenomic data mining using tensor decomposition, Bioinformatics, (2018), 1–7. |
| [10] | Y. H. Taguchi, Identification of candidate drugs using tensor-decomposition-based unsupervised feature extraction in integrated analysis of gene expression between diseases and DrugMatrix datasets, Sci. Rep., 7 (2017), 13733. |
| [11] | Y. Wang, X. Yao, K. Zhou, X. Qin, J. D. Kim, K. B Cohen and J. Xia, Guideline design of an active gene annotation corpus for the purpose of drug repurposing, 2018 11th International Congress on Image and Signal Processing, BioMedical Engineering and Informatics(CISP-BMEI 2018), Oct, 2018, Beijing. (Accepted) |
| [12] | T. G. Kolda and W. B. Bader, Tensor decompositions and applications. SIAM Rev., 51 (2009), 455–500. |
| [13] | R. Stephan, O. Shchur and S. Günnemann, Introduction to tensor decompositions and their applications in machine learning, arXiv preprint, 1711(2017),10781. |
| [14] | L. Hao, S. Liang, J. Ye and Z. Xu, TensorD: A tensor decomposition library in TensorFlow, Neurocomputing, 318(2018), 196–200. |
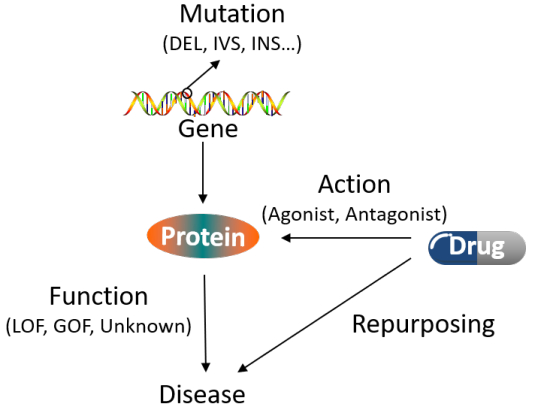
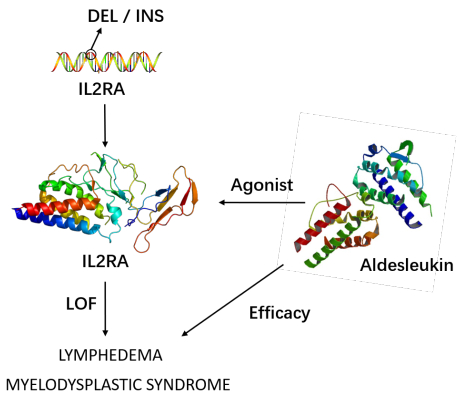
Figures(6) / Tables(5)

Kaiyin Zhou, YuxingWang, Sheng Zhang, Mina Gachloo, Jin-Dong Kim, Qi Luo, Kevin Bretonnel Cohen, Jingbo Xia. GOF/LOF knowledge inference with tensor decomposition in support of high order link discovery for gene, mutation and disease[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1376-1391. doi: 10.3934/mbe.2019067







 DownLoad:
DownLoad: