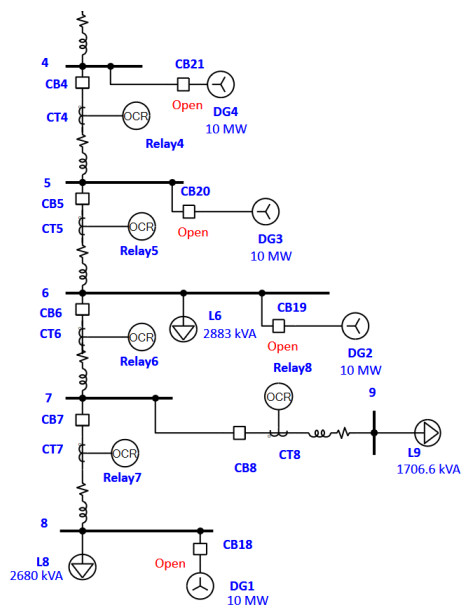
More and more distributed energy resources (DERs) are being added to the medium-voltage (MV) or low-voltage (LV) radial distribution networks (RDNs). These distributed power sources will cause the redistribution of power flow and fault current, bringing new challenges to the coordination of power system protection. An adaptive protection coordination strategy is proposed in this paper. It will trace the connectivity of the system structure to determine the set of relay numbers as a tracking path. According to the topology of the system structure, the tracking path can be divided into two categories: the main feeder path and the branch path. The time multiplier setting (TMS) of each relay can be used to evaluate the operation time of the over-current relay (OCR), and the operation time of the relay can be used to evaluate the fitness of the TMS setting combination. Furthermore, the relay protection coordination problem can be modeled to minimize the accumulated summation of all primary and backup relay operation time (OT) subject to the coordination time interval (CTI) limitation. A modified particle swarm optimization (MPSO) algorithm with adaptive self-cognition and society operation scheme (ASSOS) was proposed and utilized to determine TMS for each relay on the tracking path. A 16-bus test MV system with distributed generators (DGs) will be applied to test the adaptive protection coordination approach proposed in this paper. The results show that the proposed MPSO algorithm reduces the overall OT and relieves the impact on protection coordination settings after DG joins the system. The paper also tests and compares the proposed MPSO with other metaheuristic intelligence-based random search algorithms to prove that MPSO possesses with increased efficiency and performance.
Citation: Tung-Sheng Zhan, Chun-Lien Su, Yih-Der Lee, Jheng-Lun Jiang, Jin-Ting Yu. Adaptive OCR coordination in distribution system with distributed energy resources contribution[J]. AIMS Energy, 2023, 11(6): 1278-1305. doi: 10.3934/energy.2023058
More and more distributed energy resources (DERs) are being added to the medium-voltage (MV) or low-voltage (LV) radial distribution networks (RDNs). These distributed power sources will cause the redistribution of power flow and fault current, bringing new challenges to the coordination of power system protection. An adaptive protection coordination strategy is proposed in this paper. It will trace the connectivity of the system structure to determine the set of relay numbers as a tracking path. According to the topology of the system structure, the tracking path can be divided into two categories: the main feeder path and the branch path. The time multiplier setting (TMS) of each relay can be used to evaluate the operation time of the over-current relay (OCR), and the operation time of the relay can be used to evaluate the fitness of the TMS setting combination. Furthermore, the relay protection coordination problem can be modeled to minimize the accumulated summation of all primary and backup relay operation time (OT) subject to the coordination time interval (CTI) limitation. A modified particle swarm optimization (MPSO) algorithm with adaptive self-cognition and society operation scheme (ASSOS) was proposed and utilized to determine TMS for each relay on the tracking path. A 16-bus test MV system with distributed generators (DGs) will be applied to test the adaptive protection coordination approach proposed in this paper. The results show that the proposed MPSO algorithm reduces the overall OT and relieves the impact on protection coordination settings after DG joins the system. The paper also tests and compares the proposed MPSO with other metaheuristic intelligence-based random search algorithms to prove that MPSO possesses with increased efficiency and performance.

| [1] |
Holguin JP, Rodriguez DC, Ramos G (2020) Reverse power flow (RPF) detection and impact on protection coordination of distribution systems. IEEE Trans Ind Appl 56: 2393–2401. https://doi.org/10.1109/TIA.2020.2969640 doi: 10.1109/TIA.2020.2969640

|
| [2] |
Zeineldin HH, Mohamed YARI, Khadkikar V, et al. (2013) A protection coordination index for evaluating distributed generation impacts on protection for meshed distribution systems. IEEE Trans Smart Grid 4: 1523–1532. https://doi.org/10.1109/TSG.2013.2263745 doi: 10.1109/TSG.2013.2263745
|
| [3] |
Wan H, Li KK, Wong KP (2010) An adaptive multiagent approach to protection relay coordination with distributed generators in industrial power distribution system. IEEE Trans Ind Appl 46: 2118–2124. https://doi.org/10.1109/TIA.2010.2059492 doi: 10.1109/TIA.2010.2059492
|
| [4] |
Isherwood N, Rahman MS, Oo AMT (2017) Distribution feeder protection and reconfiguration using multi-agent approach. Proceeding of Australasian Universities Power Engineering Conference (AUPEC) 1–6. https://doi.org/10.1109/AUPEC.2017.8282425 doi: 10.1109/AUPEC.2017.8282425
|
| [5] |
Kayyali D, Zeineldin H, Diabat A, et al. (2020) An optimal integrated approach considering distribution system reconfiguration and protection coordination. Proceeding of 2020 IEEE Power & Energy Society General Meeting (PESGM) 1–5. https://doi.org/10.1109/PESGM41954.2020.9281412 doi: 10.1109/PESGM41954.2020.9281412
|
| [6] |
Ghotbi-Maleki M, Chabanloo RM, Zeineldin HH, et al. (2021) Design of setting group-based overcurrent protection scheme for active distribution networks using MILP. IEEE Trans Smart Grid 12: 1185–1193. https://doi.org/10.1109/TSG.2020.3027371 doi: 10.1109/TSG.2020.3027371
|
| [7] |
Alam MN, Chakrabarti S, Tiwari VK (2020) Protection coordination with high penetration of solar power to distribution networks. Proceeding of 2020 2nd International Conference on Smart Power & Internet Energy Systems (SPIES) 132–137. https://doi.org/10.1109/SPIES48661.2020.9243146 doi: 10.1109/SPIES48661.2020.9243146
|
| [8] |
Abdul Rahim MN, Mokhlis H, Bakar AHA, et al. (2019) Protection coordination toward optimal network reconfiguration and DG Sizing. IEEE Access 7: 163700–163718. https://doi.org/10.1109/ACCESS.2019.2952652 doi: 10.1109/ACCESS.2019.2952652
|
| [9] |
Zhan H, Wang C, Wang Y, et al. (2016) Relay protection coordination integrated optimal placement and sizing of distributed generation sources in distribution networks. IEEE Trans Smart Grid 7: 55–65. https://doi.org//10.1109/TSG.2015.2420667 doi: 10.1109/TSG.2015.2420667
|
| [10] |
Pedraza A, Reyes D, Gomez C, et al. (2015) Optimization methodology to distributed generation location in distribution networks assessing protections coordination. IEEE Latin America Trans 13: 1398–1406. https://doi.org//10.1109/TLA.2015.7111995 doi: 10.1109/TLA.2015.7111995
|
| [11] |
Saldarriaga-Zuluaga SD, López-Lezama JM, Muñoz-Galeano N (2021) Adaptive protection coordination scheme in microgrids using directional over-current relays with non-standard characteristics. Heliyon 7: e06665. https://doi.org/10.1016/j.heliyon.2021.e06665 doi: 10.1016/j.heliyon.2021.e06665
|
| [12] |
Mahat P, Chen Z, Bak-Jensen B, et al. (2011) A simple adaptive overcurrent protection of distribution systems with distributed generation. IEEE Trans Smart Grid 2: 428–437. https://doi.org//10.1109/TSG.2011.2149550 doi: 10.1109/TSG.2011.2149550
|
| [13] |
Jongepier AG, Van der Sluis L (1997) Adaptive distance protection of double-circuit lines using artificial neural networks. IEEE Trans on Power Delivery 12: 97–105. https://doi.org//10.1109/61.568229 doi: 10.1109/61.568229
|
| [14] |
Musirikare A, Pujiantara M, Tjahjono A, et al. (2018) ANN-based modeling of directional overcurrent relay characteristics applied in radial distribution system with distributed generations. Proceeding of 2018 10th International Conference on Information Technology and Electrical Engineering (ICITEE) 52–57. https://doi.org//10.1109/ICITEED.2018.8534834 doi: 10.1109/ICITEED.2018.8534834
|
| [15] |
Rahmatullah D, Dewantara BY, Iradiratu DPK (2018) Adaptive DOCR coordination in loop electrical distribution system with DG using artificial neural network LMBP. Proceeding of 2018 International Seminar on Research of Information Technology and Intelligent Systems (ISRITI) 560–565. https://doi.org//10.1109/ISRITI.2018.8864433 doi: 10.1109/ISRITI.2018.8864433
|
| [16] |
Chiang MY, Huang SC, Hsiao TC, et al. (2022) Optimal sizing and location of photovoltaic generation and energy storage systems in an unbalanced distribution system. Energies 15: 6682. https://doi.org/10.3390/en15186682 doi: 10.3390/en15186682
|
| [17] |
Javadian SAM, Tamizkar R, Haghifam MR (2009) A Protection and reconfiguration scheme for distribution networks with DG. Proceeding of 2009 IEEE Bucharest Power Tech Conference 1–8. https://doi.org/10.1109/PTC.2009.5282063 doi: 10.1109/PTC.2009.5282063
|
| [18] |
Akmal M, Al-Naemi F, Iqbal N, et al. (2019) Impact of distributed PV generation on relay coordination and power quality. Proceeding of 2019 IEEE Milan PowerTech 1–6. https://doi.org/10.1109/PTC.2019.8810791 doi: 10.1109/PTC.2019.8810791
|
| [19] |
Soni AK, Kumar A, Panda RK, et al. (2023) Adaptive coordination of relays in AC microgrid considering operational and topological changes. IEEE Systems Journal 17: 3071–3082. https://doi.org/10.1109/JSYST.2022.3227311 doi: 10.1109/JSYST.2022.3227311
|
| [20] | The Institute of Electrical and Electronics Engineers, Inc. (2001) IEEE recommended practice for protection and coordination of industrial and commercial power systems, New York: IEEE press, 1–710. |
| [21] |
Kennedy J, Eberhart R (1995) Particle swarm optimization. Proceedings of the IEEE International Conference on Neural Networks (ICNN'95) 4: 1942–1948. https://doi.org//10.1109/ICNN.1995.488968 doi: 10.1109/ICNN.1995.488968
|
| [22] |
Eberhart R, Shi Y (2000) Comparing inertia weights and constriction factors in particle swarm optimization. Proceedings of the 2000 Congress on Evolutionary Computation (CEC00) 1: 84–88. https://doi.org//10.1109/CEC.2000.870279 doi: 10.1109/CEC.2000.870279
|



















Figures(20) / Tables(9)

Tung-Sheng Zhan, Chun-Lien Su, Yih-Der Lee, Jheng-Lun Jiang, Jin-Ting Yu. Adaptive OCR coordination in distribution system with distributed energy resources contribution[J]. AIMS Energy, 2023, 11(6): 1278-1305. doi: 10.3934/energy.2023058







 DownLoad:
DownLoad: