Citation: Jacob Fosso-Tande, Cody Black, Stephen G. Aller, Lanyuan Lu, Ronald D. Hills Jr. Simulation of lipid-protein interactions with the CgProt force field[J]. AIMS Molecular Science, 2017, 4(3): 352-369. doi: 10.3934/molsci.2017.3.352

| [1] |
Barrera NP, Zhou M, Robinson CV (2013) The role of lipids in defining membrane protein interactions: Insights from mass spectrometry. Trends Cell Biol 23: 1-8. doi: 10.1016/j.tcb.2012.08.007

|
| [2] |
Smith AW (2012) Lipid-protein interactions in biological membranes: A dynamic perspective. Biochim Biophys Acta Biomembr 1818: 172-177. doi: 10.1016/j.bbamem.2011.06.015
|
| [3] |
Martinez-Seara H, Rog T, Karttunen M, et al. (2008) Influence of cis double-bond parametrization on lipid membrane properties: How seemingly insignificant details in force-field change even qualitative trends. J Chem Phys 129: 105103. doi: 10.1063/1.2976443
|
| [4] |
Ogata K, Nakamura S (2015) Improvement of parameters of the AMBER potential force field for phospholipids for description of thermal phase transitions. J Phys Chem B 119: 9726-9739. doi: 10.1021/acs.jpcb.5b01656
|
| [5] |
Venable RM, Brown FL, Pastor RW (2015) Mechanical properties of lipid bilayers from molecular dynamics simulation. Chem Phys Lipids 192: 60-74. doi: 10.1016/j.chemphyslip.2015.07.014
|
| [6] |
Paloncyova M, Fabre G, DeVane RH, et al. (2014) Benchmarking of force fields for molecule-membrane interactions. J Chem Theory Comput 10: 4143-4151. doi: 10.1021/ct500419b
|
| [7] |
Venable RM, Ingolfsson HI, Lerner MG, et al. (2017) Lipid and peptide diffusion in bilayers: The Saffman-Delbruck model and periodic boundary conditions. J Phys Chem B 121: 3443-3457. doi: 10.1021/acs.jpcb.6b09111
|
| [8] |
Perilla JR, Goh BC, Cassidy CK, et al. (2015) Molecular dynamics simulations of large macromolecular complexes. Curr Opin Struct Biol 31: 64-74. doi: 10.1016/j.sbi.2015.03.007
|
| [9] |
Baaden M, Marrink SJ (2013) Coarse-grain modelling of protein-protein interactions. Curr Opin Struct Biol 23: 878-886. doi: 10.1016/j.sbi.2013.09.004
|
| [10] |
Hills Jr RD, Brooks III CL (2009) Insights from coarse-grained Go models for protein folding and dynamics. Int J Mol Sci 10: 889-905. doi: 10.3390/ijms10030889
|
| [11] |
Hills Jr RD (2014) Balancing bond, nonbond, and Go-like terms in coarse grain simulations of conformational dynamics. Methods Mol Biol 1084: 123-140. doi: 10.1007/978-1-62703-658-0_7
|
| [12] |
Jackson J, Nguyen K, Whitford PC (2015) Exploring the balance between folding and functional dynamics in proteins and RNA. Int J Mol Sci 16: 6868-6889. doi: 10.3390/ijms16046868
|
| [13] |
Kmiecik S, Gront D, Kolinski M, et al. (2016) Coarse-grained protein models and their applications. Chem Rev 116: 7898-7936. doi: 10.1021/acs.chemrev.6b00163
|
| [14] |
Ramirez CL, Petruk A, Bringas M, et al. (2016) Coarse-grained simulations of heme proteins: Validation and study of large conformational transitions. J Chem Theory Comput 12: 3390-3397. doi: 10.1021/acs.jctc.6b00278
|
| [15] |
De Sancho D, Best RB (2012) Modulation of an IDP binding mechanism and rates by helix propensity and non-native interactions: Association of HIF1alpha with CBP. Mol Biosyst 8: 256-267. doi: 10.1039/C1MB05252G
|
| [16] |
Periole X, Cavalli M, Marrink SJ, et al. (2009) Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J Chem Theory Comput 5: 2531-2543. doi: 10.1021/ct9002114
|
| [17] |
Shen H, Moustafa IM, Cameron CE, et al. (2012) Exploring the dynamics of four RNA-dependent RNA polymerases by a coarse-grained model. J Phys Chem B 116: 14515-14524. doi: 10.1021/jp302709v
|
| [18] |
Dony N, Crowet JM, Joris B, et al. (2013) SAHBNET, an accessible surface-based elastic network: An application to membrane protein. Int J Mol Sci 14: 11510-11526. doi: 10.3390/ijms140611510
|
| [19] |
Shimamura T, Weyand S, Beckstein O, et al. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328: 470-473. doi: 10.1126/science.1186303
|
| [20] |
Ward AB, Guvench O, Hills Jr RD (2012) Coarse grain lipid-protein molecular interactions and diffusion with MsbA flippase. Proteins 80: 2178-2190. doi: 10.1002/prot.24108
|
| [21] |
Prasanna X, Sengupta D, Chattopadhyay A (2016) Cholesterol-dependent conformational plasticity in GPCR dimers. Sci Rep 6: 31858. doi: 10.1038/srep31858
|
| [22] |
Poyry S, Vattulainen I (2016) Role of charged lipids in membrane structures: Insight given by simulations. Biochim Biophys Acta Biomembr 1858: 2322-2333. doi: 10.1016/j.bbamem.2016.03.016
|
| [23] |
Hedger G, Sansom MS (2016) Lipid interaction sites on channels, transporters and receptors: Recent insights from molecular dynamics simulations. Biochim Biophys Acta Biomembr 1858: 2390-2400. doi: 10.1016/j.bbamem.2016.02.037
|
| [24] |
Provasi D, Boz MB, Johnston JM, et al. (2015) Preferred supramolecular organization and dimer interfaces of opioid receptors from simulated self-association. PLOS Comput Biol 11: e1004148. doi: 10.1371/journal.pcbi.1004148
|
| [25] |
Kalli AC, Sansom MS, Reithmeier RA (2015) Molecular dynamics simulations of the bacterial UraA H+-uracil symporter in lipid bilayers reveal a closed state and a selective interaction with cardiolipin. PLOS Comput Biol 11: e1004123. doi: 10.1371/journal.pcbi.1004123
|
| [26] |
Marrink SJ, Risselada HJ, Yefimov S, et al. (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B 111: 7812-7824. doi: 10.1021/jp071097f
|
| [27] |
de Jong DH, Periole X, Marrink SJ (2012) Dimerization of amino acid side chains: Lessons from the comparison of different force fields. J Chem Theory Comput 8: 1003-1014. doi: 10.1021/ct200599d
|
| [28] |
Singh G, Tieleman DP (2011) Using the Wimley-White hydrophobicity scale as a direct quantitative test of force fields: The MARTINI coarse-grained model. J Chem Theory Comput 7: 2316-2324. doi: 10.1021/ct2002623
|
| [29] |
Stark AC, Andrews CT, Elcock AH (2013) Toward optimized potential functions for protein-protein interactions in aqueous solutions: Osmotic second virial coefficient calculations using the MARTINI coarse-grained force field. J Chem Theory Comput 9: 4176-4185. doi: 10.1021/ct400008p
|
| [30] |
Bereau T, Kremer K (2016) Protein-backbone thermodynamics across the membrane interface. J Phys Chem B 120: 6391-6400. doi: 10.1021/acs.jpcb.6b03682
|
| [31] |
Bereau T, Bennett WF, Pfaendtner J, et al. (2015) Folding and insertion thermodynamics of the transmembrane WALP peptide. J Chem Phys 143: 243127. doi: 10.1063/1.4935487
|
| [32] | Rodgers JM, Sorensen J, de Meyer FJ, et al. (2012) Understanding the phase behavior of coarse-grained model lipid bilayers through computational calorimetry. J Phys Chem B 116: 1551-1569. |
| [33] |
Arnarez C, Uusitalo JJ, Masman MF, et al. (2015) Dry Martini, a coarse-grained force field for lipid membrane simulations with implicit solvent. J Chem Theory Comput 11: 260-275. doi: 10.1021/ct500477k
|
| [34] |
Ingolfsson HI, Melo MN, van Eerden FJ, et al. (2014) Lipid organization of the plasma membrane. J Am Chem Soc 136: 14554-14559. doi: 10.1021/ja507832e
|
| [35] |
Melo MN, Ingolfsson HI, Marrink SJ (2015) Parameters for Martini sterols and hopanoids based on a virtual-site description. J Chem Phys 143: 243152. doi: 10.1063/1.4937783
|
| [36] |
Paloncyova M, Vavrova K, Sovova Z, et al. (2015) Structural changes in ceramide bilayers rationalize increased permeation through stratum corneum models with shorter acyl tails. J Phys Chem B 119: 9811-9819. doi: 10.1021/acs.jpcb.5b05522
|
| [37] |
Periole X, Marrink SJ (2013) The Martini coarse-grained force field. Methods Mol Biol 924: 533-565. doi: 10.1007/978-1-62703-017-5_20
|
| [38] |
Marrink SJ, Tieleman DP (2013) Perspective on the Martini model. Chem Soc Rev 42: 6801-6822. doi: 10.1039/c3cs60093a
|
| [39] |
Baron R, de Vries AH, Hunenberger PH, et al. (2006) Comparison of atomic-level and coarse-grained models for liquid hydrocarbons from molecular dynamics configurational entropy estimates. J Phys Chem B 110: 8464-8473. doi: 10.1021/jp055888y
|
| [40] |
Hills Jr RD, Lu L, Voth GA (2010) Multiscale coarse-graining of the protein energy landscape. PLOS Comput Biol 6: e1000827. doi: 10.1371/journal.pcbi.1000827
|
| [41] |
Jia Z, Chen J (2016) Necessity of high-resolution for coarse-grained modeling of flexible proteins. J Comput Chem 37: 1725-1733. doi: 10.1002/jcc.24391
|
| [42] |
Miguel V, Perillo MA, Villarreal MA (2016) Improved prediction of bilayer and monolayer properties using a refined BMW-MARTINI force field. Biochim Biophys Acta Biomembr 1858: 2903-2910. doi: 10.1016/j.bbamem.2016.08.016
|
| [43] |
Wu Z, Cui Q, Yethiraj A (2010) A new coarse-grained model for water: The importance of electrostatic interactions. J Phys Chem B 114: 10524-10529. doi: 10.1021/jp1019763
|
| [44] |
Wu Z, Cui Q, Yethiraj A (2011) A new coarse-grained force field for membrane-peptide simulations. J Chem Theory Comput 7: 3793-3802. doi: 10.1021/ct200593t
|
| [45] |
Yesylevskyy SO, Schafer LV, Sengupta D, et al. (2010) Polarizable water model for the coarse-grained MARTINI force field. PLOS Comput Biol 6: e1000810. doi: 10.1371/journal.pcbi.1000810
|
| [46] |
de Jong DH, Singh G, Bennett WFD, et al. (2013) Improved parameters for the Martini coarse-grained protein force field. J Chem Theory Comput 9: 687-697. doi: 10.1021/ct300646g
|
| [47] |
Lu L, Voth GA (2009) Systematic coarse-graining of a multicomponent lipid bilayer. J Phys Chem B 113: 1501-1510. doi: 10.1021/jp809604k
|
| [48] |
Wang ZJ, Deserno M (2010) A systematically coarse-grained solvent-free model for quantitative phospholipid bilayer simulations. J Phys Chem B 114: 11207-11220. doi: 10.1021/jp102543j
|
| [49] |
Han W, Schulten K (2012) Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: Improved backbone hydration and interactions between charged side chains. J Chem Theory Comput 8: 4413-4424. doi: 10.1021/ct300696c
|
| [50] |
Han W, Wan CK, Wu YD (2008) Toward a coarse-grained protein model coupled with a coarse-grained solvent model: Solvation free energies of amino acid side chains. J Chem Theory Comput 4: 1891-1901. doi: 10.1021/ct800184c
|
| [51] |
Ganesan SJ, Matysiak S (2014) Role of backbone dipole interactions in the formation of secondary and supersecondary structures of proteins. J Chem Theory Comput 10: 2569-2576. doi: 10.1021/ct401087a
|
| [52] | Noid WG, Chu JW, Ayton GS, et al. (2008) The multiscale coarse-graining method. I. A rigorous bridge between atomistic and coarse-grained models. J Chem Phys 128: 244114. |
| [53] |
Noid WG, Chu JW, Ayton GS, et al. (2007) Multiscale coarse-graining and structural correlations: Connections to liquid-state theory. J Phys Chem B 111: 4116-4127. doi: 10.1021/jp068549t
|
| [54] | Pall S, Abraham MJ, Kutzner C, et al. (2015) Tackling exascale software challenges in molecular dynamics simulations with GROMACS. Proc EASC 2015 LNCS 8759: 3-27. |
| [55] |
Hills Jr RD, McGlinchey N (2016) Model parameters for simulation of physiological lipids. J Comput Chem 37: 1112-1118. doi: 10.1002/jcc.24324
|
| [56] |
Monticelli L, Kandasamy SK, Periole X, et al. (2008) The MARTINI coarse-grained force field: Extension to proteins. J Chem Theory Comput 4: 819-834. doi: 10.1021/ct700324x
|
| [57] |
Bond PJ, Wee CL, Sansom MS (2008) Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry 47: 11321-11331. doi: 10.1021/bi800642m
|
| [58] | Winger M, Trzesniak D, Baron R, et al. (2009) On using a too large integration time step in molecular dynamics simulations of coarse-grained molecular models. Phys Chem Chem Phys 11: 1934-1941. |
| [59] | Karanicolas J, Brooks III CL (2002) The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci 11: 2351-2361. |
| [60] |
Bereau T, Deserno M (2009) Generic coarse-grained model for protein folding and aggregation. J Chem Phys 130: 235106. doi: 10.1063/1.3152842
|
| [61] |
Herzog FA, Braun L, Schoen I, et al. (2016) Improved side chain dynamics in MARTINI simulations of protein-lipid interfaces. J Chem Theory Comput 12: 2446-24458. doi: 10.1021/acs.jctc.6b00122
|
| [62] |
Cerutti DS, Duke R, Freddolino PL, et al. (2008) Vulnerability in popular molecular dynamics packages concerning Langevin and Andersen dynamics. J Chem Theory Comput 4: 1669-1680. doi: 10.1021/ct8002173
|
| [63] |
Bereau T, Wang ZJ, Deserno M (2014) More than the sum of its parts: Coarse-grained peptide-lipid interactions from a simple cross-parametrization. J Chem Phys 140: 115101. doi: 10.1063/1.4867465
|
| [64] |
Bond PJ, Holyoake J, Ivetac A, et al. (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol 157: 593-605. doi: 10.1016/j.jsb.2006.10.004
|
| [65] |
Kar P, Gopal SM, Cheng YM, et al. (2014) Transferring the PRIMO coarse-grained force field to the membrane environment: Simulations of membrane proteins and helix-helix association. J Chem Theory Comput 10: 3459-3472. doi: 10.1021/ct500443v
|
| [66] |
Hall BA, Chetwynd AP, Sansom MS (2011) Exploring peptide-membrane interactions with coarse-grained MD simulations. Biophys J 100: 1940-1948. doi: 10.1016/j.bpj.2011.02.041
|
| [67] |
Kim T, Im W (2010) Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys J 99: 175-183. doi: 10.1016/j.bpj.2010.04.015
|
| [68] |
Liu F, Lewis RN, Hodges RS, et al. (2004) Effect of variations in the structure of a polyleucine-based alpha-helical transmembrane peptide on its interaction with phosphatidylethanolamine bilayers. Biophys J 87: 2470-2482. doi: 10.1529/biophysj.104.046342
|
| [69] |
Lear JD, Wasserman ZR, DeGrado WF (1988) Synthetic amphiphilic peptide models for protein ion channels. Science 240: 1177-1181. doi: 10.1126/science.2453923
|
| [70] |
Li J, Jaimes KF, Aller SG (2014) Refined structures of mouse P-glycoprotein. Protein Sci 23: 34-46. doi: 10.1002/pro.2387
|
| [71] |
Costa JA, Nguyen DA, Leal-Pinto E, et al. (2013) Wicking: A rapid method for manually inserting ion channels into planar lipid bilayers. PLOS ONE 8: e60836. doi: 10.1371/journal.pone.0060836
|
| [72] |
Javanainen M (2014) Universal method for embedding proteins into complex lipid bilayers for molecular dynamics simulations. J Chem Theory Comput 10: 2577-2582. doi: 10.1021/ct500046e
|
| [73] |
Daily MD, Olsen BN, Schlesinger PH, et al. (2014) Improved coarse-grained modeling of cholesterol-containing lipid bilayers. J Chem Theory Comput 10: 2137-2150. doi: 10.1021/ct401028g
|
| [74] |
Nguyen TH, Liu Z, Moore PB (2013) Molecular dynamics simulations of homo-oligomeric bundles embedded within a lipid bilayer. Biophys J 105: 1569-1580. doi: 10.1016/j.bpj.2013.07.053
|
| [75] |
Holt A, Koehorst RB, Rutters-Meijneke T, et al. (2009) Tilt and rotation angles of a transmembrane model peptide as studied by fluorescence spectroscopy. Biophys J 97: 2258-2266. doi: 10.1016/j.bpj.2009.07.042
|
| [76] |
Ozdirekcan S, Etchebest C, Killian JA, et al. (2007) On the orientation of a designed transmembrane peptide: Toward the right tilt angle? J Am Chem Soc 129: 15174-15181. doi: 10.1021/ja073784q
|
| [77] |
Moeller A, Lee SC, Tao H, et al. (2015) Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 23: 450-460. doi: 10.1016/j.str.2014.12.013
|
| [78] |
Ward AB, Reyes CL, Yu J, et al. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci USA 104: 19005-19010. doi: 10.1073/pnas.0709388104
|
| [79] |
Pan L, Aller SG (2015) Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics. Sci Rep 5: 7880. doi: 10.1038/srep07880
|
| [80] |
Loo TW, Clarke DM (2000) Drug-stimulated ATPase activity of human P-glycoprotein is blocked by disulfide cross-linking between the nucleotide-binding sites. J Biol Chem 275: 19435-19438. doi: 10.1074/jbc.C000222200
|
| [81] |
Urbatsch IL, Gimi K, Wilke-Mounts S, et al. (2001) Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J Biol Chem 276: 26980-26987. doi: 10.1074/jbc.M010829200
|
| [82] |
East JM, Melville D, Lee AG (1985) Exchange rates and numbers of annular lipids for the calcium and magnesium ion dependent adenosinetriphosphatase. Biochemistry 24: 2615-2623. doi: 10.1021/bi00332a005
|
| [83] |
Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. J Mol Graph 14: 33-38. doi: 10.1016/0263-7855(96)00018-5
|
| [84] |
Bechara C, Noll A, Morgner N, et al. (2015) A subset of annular lipids is linked to the flippase activity of an ABC transporter. Nat Chem 7: 255-262. doi: 10.1038/nchem.2172
|
| [85] |
Aller SG, Yu J, Ward AB, et al. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323: 1718-1722. doi: 10.1126/science.1168750
|
| [86] |
Habeck M, Haviv H, Katz A, et al. (2015) Stimulation, inhibition, or stabilization of Na,K-ATPase caused by specific lipid interactions at distinct sites. J Biol Chem 290: 4829-4842. doi: 10.1074/jbc.M114.611384
|
| [87] |
Kucerka N, van Oosten B, Pan J, et al. (2015) Molecular structures of fluid phosphatidylethanolamine bilayers obtained from simulation-to-experiment comparisons and experimental scattering density profiles. J Phys Chem B 119: 1947-1956. doi: 10.1021/jp511159q
|
| [88] |
Gleason NJ, Vostrikov VV, Greathouse DV, et al. (2013) Buried lysine, but not arginine, titrates and alters transmembrane helix tilt. Proc Natl Acad Sci USA 110: 1692-1695. doi: 10.1073/pnas.1215400110
|
| [89] |
Hu Y, Sinha SK, Patel S (2014) Reconciling structural and thermodynamic predictions using all-atom and coarse-grain force fields: The case of charged oligo-arginine translocation into DMPC bilayers. J Phys Chem B 118: 11973-11992. doi: 10.1021/jp504853t
|
| [90] |
Sun D, Forsman J, Woodward CE (2015) Evaluating force fields for the computational prediction of ionized arginine and lysine side-chains partitioning into lipid bilayers and octanol. J Chem Theory Comput 11: 1775-1791. doi: 10.1021/ct501063a
|
| [91] |
MacCallum JL, Bennett WF, Tieleman DP (2008) Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J 94: 3393-3404. doi: 10.1529/biophysj.107.112805
|
| [92] |
Matalon E, Kaminker I, Zimmermann H, et al. (2013) Topology of the trans-membrane peptide WALP23 in model membranes under negative mismatch conditions. J Phys Chem B 117: 2280-2293. doi: 10.1021/jp310056h
|
| [93] |
Clay AT, Lu P, Sharom FJ (2015) Interaction of the p-glycoprotein multidrug transporter with sterols. Biochemistry 54: 6586-6597. doi: 10.1021/acs.biochem.5b00904
|
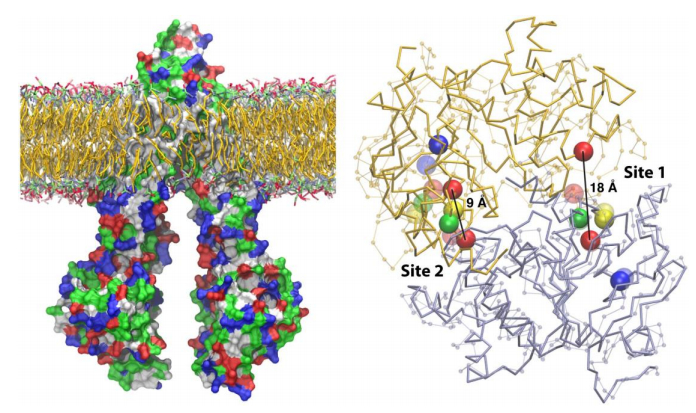
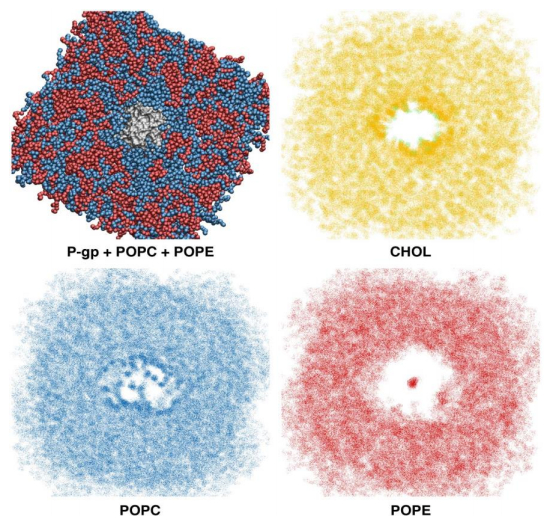
Figures(6) / Tables(3)

Jacob Fosso-Tande, Cody Black, Stephen G. Aller, Lanyuan Lu, Ronald D. Hills Jr. Simulation of lipid-protein interactions with the CgProt force field[J]. AIMS Molecular Science, 2017, 4(3): 352-369. doi: 10.3934/molsci.2017.3.352







 DownLoad:
DownLoad: