Citation: Ignacio Alvarez-Castro, Jarad Niemi. Fully Bayesian analysis of allele-specific RNA-seq data[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 7751-7770. doi: 10.3934/mbe.2019389

| [1] | S. Datta and D. Nettleton, Statistical Analysis of Next Generation Sequencing Data, Springer, 2014. Available from: http://link.springer.com/content/pdf/10.1007/978-3-319-07212-8.pdf. |
| [2] | W. Sun and Y. Hu, Mapping of expression quantitative trait loci using RNA-seq data, in Statistical Analysis of Next Generation Sequencing Data (eds. D. Nettleton and S. Datta), 2014, 25–50. |
| [3] | P. S. Schnable and N. M. Springer, Progress toward understanding heterosis in crop plants, Annu. Rev. Plant Biol., 64 (2013), 71–88. |
| [4] | A. Paschold, Y. Jia, C. Marcon, et al., Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents., Genome Res., 22 (2012), 2445–2454. |
| [5] | G. D. M. Bell, N. C. Kane, L. H. Rieseberg, et al., RNA-Seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations, Genome Biol. Evol., 5 (2013), 1309–1323. |
| [6] | J. K. Pickrell, J. C. Marioni, A. A. Pai, et al., Understanding mechanisms underlying human gene expression variation with rna sequencing, Nature, 464 (2010), 768–772. |
| [7] | W. Sun and Y. Hu, eQTL Mapping Using RNA-seq Data, Stat. Biosci., 5 (2013), 198–219. |
| [8] | C. T. Harvey, G. A. Moyerbrailean, G. O. Davis, et al., Quasar: quantitative allele-specific analysis of reads, Bioinformatics, 31 (2014), 1235–1242. |
| [9] | N. Raghupathy, K. Choi, M. J. Vincent, et al., Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression, Bioinformatics, 34 (2018), 2177–2184. |
| [10] | S. Srivastava and L. Chen, A two-parameter generalized Poisson model to improve the analysis of RNA-seq data., Nucleic Acids Res., 38 (2010), e170. |
| [11] | X. Wei and X. Wang, A computational workflow to identify allele-specific expression and epigenetic modification in maize., Genom. Proteom. Bioinf., 11 (2013), 247–252. |
| [12] | M. D. Robinson, D. J. McCarthy and G. K. Smyth, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data., Bioinformatics (Oxford, England), 26 (2010), 139–140. |
| [13] | D. J. Lorenz, R. S. Gill, R. Mitra, et al., Using RNA-seq Data to Detect Differentially Expressed Genes, in Statistical Analysis of Next Generation Sequencing Data (eds. S. Datta and D. Nettleton), 2014, chapter 2, 25–49. |
| [14] | Y.-J. Hu, W. Sun, J.-Y. Tzeng, et al., Proper use of allele-specific expression improves statistical power for cis -eQTL mapping with RNA-seq data, J. Am. Stat. Assoc., 110 (2015), 962–974. |
| [15] | W. Landau, J. Niemi and D. Nettleton, Fully bayesian analysis of rna-seq counts for the detection of gene expression heterosis, J. Am. Stat. Assoc., 114 (2019), 601–612. |
| [16] | N. I. Panousis, M. Gutierrez-Arcelus, E. T. Dermitzakis, et al., Allelic mapping bias in RNA-sequencing is not a major confounder in eQTL studies, Genome. Biol., 15 (2014), 467. |
| [17] | J. F. Degner, J. C. Marioni, A. A. Pai, et al., Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data, Bioinformatics, 25 (2009), 3207–3212. |
| [18] | R. Vijaya Satya, N. Zavaljevski and J. Reifman, A new strategy to reduce allelic bias in RNA-Seq readmapping, Nucleic Acids Res., 40 (2012), 1–9. |
| [19] | K. R. Stevenson, J. D. Coolon and P. J. Wittkopp, Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome., BMC Genom., 14 (2013), 536. |
| [20] | Y. Chen, A. T. L. Lun and G. K. Smyth, Differential expression analysis of complex RNA-seq experiments using edgeR, in Statistical Analysis of Next Generation Sequencing Data, Springer, Cham, 2014, 51–74. |
| [21] | T. Park and G. Casella, The Bayesian lasso, J. Am. Stat. Assoc., 103 (2008), 681–686. |
| [22] | C. M. Carvalho, N. G. Polson and J. G. Scott, Handling Sparsity via the Horseshoe, J. Mach. Learn. Res., 5 (2009), 73–80. |
| [23] | A. Gelman, Prior distributions for variance parameters in hierarchical models, Bayesian Analysis, 1 (2006), 515–533. |
| [24] | A. Gelman, J. B. Carlin, H. S. Stern,et al., Bayesian Data Analysis, CRC press, 2013. |
| [25] | J. K. Ghosh, M. Delampady and T. Samanta, An Introduction to Bayesian Analysis, Springer, 2006. Available from: http://onlinelibrary.wiley.com/doi/10.1002/9781118684818.ch16/summary. |
| [26] | L. G. León-Novelo, L. M. McIntyre, J. M. Fear, et al., A flexible Bayesian method for detecting allelic imbalance in RNA-seq data, BMC Genom., 15 (2014), 920. |
| [27] | J. Niemi, E. Mittman, W. Landau, et al., Empirical Bayes analysis of RNA-seq data for detection of gene expression heterosis, J. Agr. Biol. Envir. St., 20 (2015), 614–628. |
| [28] | M. A. Van De Wiel, G. G. R. Leday, L. Pardo, et al., Bayesian analysis of RNA sequencing data by estimating multiple shrinkage priors, Biostatistics, 14 (2013), 113–128. |
| [29] | W. Landau and J. Niemi, A fully Bayesian strategy for high-dimensional hierarchical modeling using massively parallel computing, 2016. Available from: http://arxiv.org/abs/1606.06659. |
| [30] | A. Lithio and D. Nettleton, Hierarchical modeling and differential expression analysis for RNA-seq experiments with inbred and hybrid genotypes, J. Agr. Biol. Envir. St., 20 (2015), 598–613. |
| [31] | M. Ventrucci, E. M. Scott and D. Cocchi, Multiple testing on standardized mortality ratios: a Bayesian hierarchical model for FDR estimation, Biostatistics, 12 (2011), 51–67. |
| [32] | P. Muller, G. Parmigiani and K. Rice, FDR and Bayesian multiple comparisons ules, 2006. Available from: http://biostats.bepress.com/jhubiostat/paper115. |
| [33] | H. Y. Bar, J. G. Booth and M. T. Wells, A bivariate model for simultaneous testing in bioinformatics data, J. Am. Stat. Assoc., 109 (2014), 537–547. |
| [34] | P. Müller, G. Parmigiani, C. Robert, et al., Optimal sample size for multiple testing: the case of gene expression microarrays, J. Am. Stat. Assoc., 99 (2004), 990–1001. |
| [35] | S. Anders and W. Huber, Differential expression analysis for sequence count data, Genome Biol., 11 (2010), R106. |
| [36] | P. R. Hahn and J. He, Elliptical slice sampling for Bayesian shrinkage regression with applications to causal inference, 2016. Available from: http://faculty.chicagobooth.edu/richard. hahn/JCGS_submit.pdf. |
| [37] | M. C. Sachs, plotROC: A tool for plotting roc curves, J. Stat. Software, 79 (2017), 1–19. |
| [38] | Y. Benjamini and Y. Hochberg, Controlling the false discovery rate: A practical and powerful approach to multiple testing, J. R. Stat. Soc-B, 57 (1995), 289–300. |
| [39] | P. S. Schnable, D. Ware, R. S. Fulton, et al., The B73 maize genome: complexity, diversity, and dynamics, Science, 326 (2009), 1112–1115. |
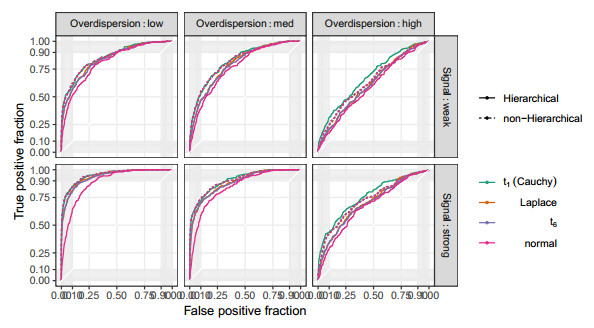
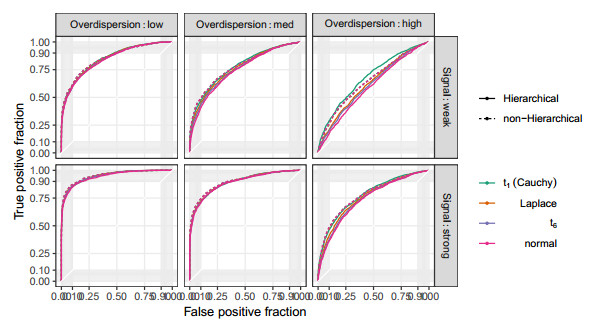
Figures(10) / Tables(2)

Ignacio Alvarez-Castro, Jarad Niemi. Fully Bayesian analysis of allele-specific RNA-seq data[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 7751-7770. doi: 10.3934/mbe.2019389







 DownLoad:
DownLoad: