Citation: Xuhua Xia. Starless bias and parameter-estimation bias in the likelihood-based phylogenetic method[J]. AIMS Genetics, 2018, 5(4): 212-223. doi: 10.3934/genet.2018.4.212

| [1] | Saitou N, Nei M (1987) The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425. |
| [2] | Desper R, Gascuel O (2004) Theoretical foundation of the balanced minimum evolution method of phylogenetic inference and its relationship to weighted least-squares tree fitting. Mol Biol Evol 21: 587–598. |
| [3] | Xia X (2014) Phylogenetic bias in the likelihood method caused by missing data coupled with Among-Site rate variation: An analytical approach. In: Basu M, Pan Y, Wang J, editors. Bioinformatics Research and Applications: Springer, 12–23. |
| [4] | Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN, editor. Mammalian Protein Metabolism. New York: Academic Press, 21–123. |
| [5] |
Hasegawa M, Kishino H (1989) Heterogeneity of tempo and mode of mitochondrial DNA evolution among mammalian orders. Jpn J Genet 64: 243–258. doi: 10.1266/jjg.64.243

|
| [6] |
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J Mol Evol 29: 170–179. doi: 10.1007/BF02100115
|
| [7] |
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174. doi: 10.1007/BF02101694
|
| [8] | Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526. |
| [9] |
Lanave C, Preparata G, Saccone C, et al. (1984) A new method for calculating evolutionary substitution rates. J Mol Evol 20: 86–93. doi: 10.1007/BF02101990
|
| [10] | Tavaré S (1986) Some probabilistic and statistical problems in the analysis of DNA sequences. In: Miura RM, editor. Lectures on Mathematics in the Life Sciences. Providence, RI: Amer Math Soc: 57–86. |
| [11] | Xia X (2017) Deriving transition probabilities and evolutionary distances from substitution rate matrix by probability reasoning. J Genet Genome Res 3: 031. |
| [12] | Xia X (2018) Nucleotide substitution models and evolutionary distances. Bioinf Cell: 269–314. |
| [13] |
Yang Z (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. doi: 10.1093/molbev/msm088
|
| [14] | Xia X (2018) Maximum likelihood in molecular phylogenetics. Bioinf Cell: 381–395. |
| [15] |
Guindon S, Dufayard JF, Lefort V, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol 59: 307–321. doi: 10.1093/sysbio/syq010
|
| [16] |
Xia X (2018) DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol 35: 1550–1552. doi: 10.1093/molbev/msy073
|
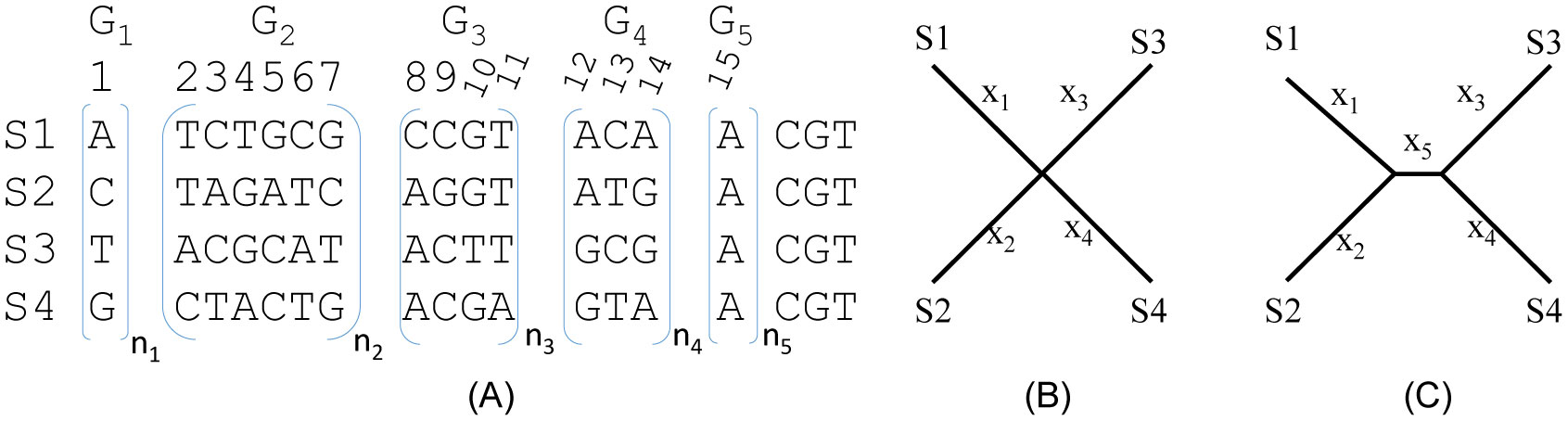

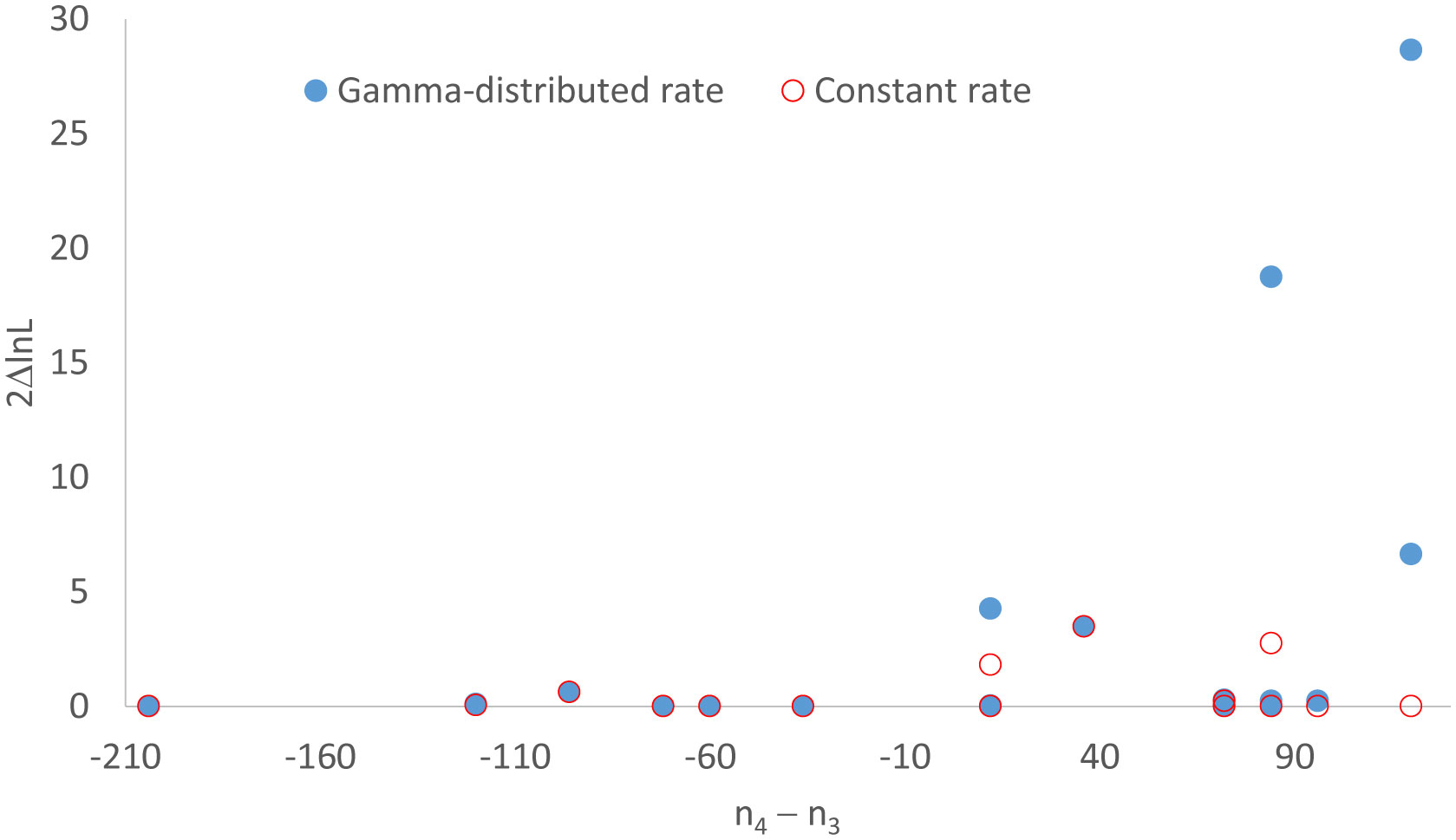
Figures(3) / Tables(2)

Xuhua Xia. Starless bias and parameter-estimation bias in the likelihood-based phylogenetic method[J]. AIMS Genetics, 2018, 5(4): 212-223. doi: 10.3934/genet.2018.4.212







 DownLoad:
DownLoad: