Citation: Natalia Ros, Laura Lomba, Ma Pilar Ribate, Estefania Zuriaga, Cristina B. García, Beatriz Giner. Acute lethal and sublethal effects of diltiazem and doxepin for four aquatic environmental bioindicators covering the trophic chain[J]. AIMS Environmental Science, 2018, 5(4): 229-243. doi: 10.3934/environsci.2018.4.229

| [1] | Abreu MS, Giacomini ACV, Gusso D, et al. (2016) Acute exposure to waterborne psychoactive drugs attract zebrafish. Comp Biochem Phys C 179: 37–43. |
| [2] |
Gros M, Petrovic M, Ginebreda A, et al. (2010) Removal of pharmaceuticals during wastewater treatment and environmental risk assessment using hazard indexes. Environ Int 36: 15–26. doi: 10.1016/j.envint.2009.09.002

|
| [3] | Budzinski H, Togola A (2006) Presence des residus de medicaments dans les differents compartiments du milieu aquatique. Environ Risque Sante 5: 248–253. |
| [4] | Haguenoer MJ, Rouban A, Aurousseau M, et. al., Médicaments et environment, Académie Nationale de Pharmacie, 2008. Available from: http://www.acadpharm.org/dos_public/1_Rapport_Med_Env_version_JMH_def_JPC.pdf. |
| [5] |
Cedergreen N, Christensen AM, Kamper A, et al. (2008) A review of independent action compared to concentration addition as reference models for mixtures of compounds with different molecular target sites. Environ Toxicol Chem 27: 1621–1632. doi: 10.1897/07-474.1
|
| [6] |
Cleuvers M (2004) Mixture toxicity of the anti-inflammatory drugs diclofenac, ibuprofen, naproxen, and acetylsalicylic acid. Ecotox Environ Safe 59: 309–315. doi: 10.1016/S0147-6513(03)00141-6
|
| [7] |
Fent K, Weston AA, Caminada D (2006) Ecotoxicology of human pharmaceuticals. Aquat Toxicol 76: 122–159. doi: 10.1016/j.aquatox.2005.09.009
|
| [8] |
Ferrari B, Paxeus N, Giudice RL, et al. (2003) Ecotoxicological impact of pharmaceuticals found in treated wastewaters: study of carbamazepine, clofibric acid, and diclofenac. Ecotox Environ Safe 55: 359–370. doi: 10.1016/S0147-6513(02)00082-9
|
| [9] |
Boxall AB, Rudd MA, Brooks BW, et al. (2012) Pharmaceuticals and Personal Care Products in the Environment: What Are the Big Questions? Environ Health Persp 120: 1221–1229. doi: 10.1289/ehp.1104477
|
| [10] |
Farre ML, Ferrer I, Ginebreda A, et al. (2001) Determination of drugs in surface water and wastewater samples by liquid chromatography-mass spectrometry: methods and preliminary results including toxicity studies with Vibrio fischeri. J Chromatogr A 938: 187–197. doi: 10.1016/S0021-9673(01)01154-2
|
| [11] |
Garcia-Galan MJ, Garrido T, Fraile J, et al. (2010) Simultaneous occurrence of nitrates and sulfonamide antibiotics in two ground water bodies of Catalonia (Spain). J Hydrol 383: 93–101. doi: 10.1016/j.jhydrol.2009.06.042
|
| [12] |
Hummel D, Loeffler D, Fink G, et al. (2006) Simultaneous determination of psychoactive drugs and their metabolites in aqueous matrices by liquid chromatography mass spectrometry. Environ Sci Technol 40: 7321–7328. doi: 10.1021/es061740w
|
| [13] |
Kolodziejska M, Maszkowska J, Bialk-Bielinska A, et al. (2013) Aquatic toxicity of four veterinary drugs commonly applied in fish farming and animal husbandry. Chemosphere 92: 1253–1259. doi: 10.1016/j.chemosphere.2013.04.057
|
| [14] |
Watanabe H, Tamura I, Abe R, et. al (2016) Chronic toxicity of an environmentally relevant mixture of pharmaceuticals to three aquatic organisms (alga, daphnid, and fish). Environ Toxicol Chem 35: 996–1006. doi: 10.1002/etc.3285
|
| [15] |
van den Brandhof EJ, Montforts M (2010) Fish embryo toxicity of carbamazepine, diclofenac and metoprolol. Ecotox Environ Safe 73: 1862–1866. doi: 10.1016/j.ecoenv.2010.08.031
|
| [16] |
Wu M, Xiang J, Que J, Chen F, Xu G (2015) Occurrence and fate of psychiatric pharmaceuticals in the urban water system of Shanghai, China. Chemosphere 138: 486–493. doi: 10.1016/j.chemosphere.2015.07.002
|
| [17] |
Dulio V, Slobodnik J (2009) NORMAN-network of reference laboratories, research centres and related organisations for monitoring of emerging environmental substances. Environ Sci Pollut R 16: S132–S135. doi: 10.1007/s11356-009-0129-1
|
| [18] |
Conley JM, Symes SJ, Schorr MS, et al. (2008) Spatial and temporal analysis of pharmaceutical concentrations in the upper Tennessee River basin. Chemosphere 73: 1178–1187. doi: 10.1016/j.chemosphere.2008.07.062
|
| [19] | Commission Regulation 65 (EC) No 1488/94. Commission Regulation 65 (EC) No 1488/94, 1994. On risk assessment of existing substances (Parts I, II, III and IV). EC catalogue numbers CR-48-96-001, 002, 003, 004-EN-C., No. 1488/94. |
| [20] |
Straub JO (2013) An environmental Risk Assessment for human-use trimethoprim in european surface waters. Antibiotics 2: 115–162. doi: 10.3390/antibiotics2010115
|
| [21] |
Sanderson H, Johnson DJ, Reitsma T, et al. (2004) Ranking and prioritizaiton of environmental risks of pharmaceuticals in surface waters. Regul Toxicol Pharm 39: 158–183. doi: 10.1016/j.yrtph.2003.12.006
|
| [22] | Mansour F, Al-Hindi M, Saad W, et al. (2016) Environmental risk analysis and priorization of pharmaceuticals in a developing world context. Sci Total Environ 557–558: 31–43. |
| [23] | Brayfield A (2014) Martindale: the complete drug reference. PhP, Pharmaceutical Press, London, UK. |
| [24] |
O'Connor SE, Grosset A, Janiak P (1999) The pharmacological basis and pathophysiological significance of the heart rate-lowering property of diltiazem. Fundam Clin Pharmacol 13: 145–153. doi: 10.1111/j.1472-8206.1999.tb00333.x
|
| [25] |
Choi K, Kim Y, Park J, et al. (2008) Seasonal variations of several pharmaceutical residues in surface water and sewage treatment plants of Han River, Korea. Sci Total Environ 405: 120–128. doi: 10.1016/j.scitotenv.2008.06.038
|
| [26] |
Azuma T, Nakada N, Yamashita N, et al. (2015) Evaluation of concentrations of pharmaceuticals detected in sewage influents in Japan by using annual shipping and sales data. Chemosphere 138: 770–776. doi: 10.1016/j.chemosphere.2015.07.073
|
| [27] |
Scott WC, Du B, Haddad SP, et al. (2016) Predicted and observed therapeutic dose exceedances of ionizable pharmaceuticals in fish plasma from urban coastal systems. Environ Toxicol Chem 35: 983–995. doi: 10.1002/etc.3236
|
| [28] |
Ellison CM, Madden JC, Judson P, et al. (2010) Using in silico tools in a weight of evidence approach to aid toxicological assessment. Mol Inform 29: 97–110. doi: 10.1002/minf.200900006
|
| [29] |
Zuriaga E, Lomba L, Royo FM, et al. (2014) Aggregation behaviour of betablocker drugs in aqueous media. New J Chem 38: 4141–4148. doi: 10.1039/C4NJ00112E
|
| [30] | UNE-EN-ISO 11348-3 (2007) Water quality-Determination of the inhibitory effect of water samples on the light emission of Vibrio fischeri (Luminescent bacteria test). |
| [31] |
Jennings VLK, Rayner-Brandes MH, Bird DJ (2001) Assessing chemical toxicity with the bioluminescent photobacterium (Vibrio fischeri): A comparison of three commercial systems. Water Res 35: 3448–3456. doi: 10.1016/S0043-1354(01)00067-7
|
| [32] | OC SE TG 202 (2004) (European C 2 method as described in the EU Regulation 440/2008). |
| [33] | OECD 202 (1984) Guideline for Testing of Chemicals No. 202. Daphnia sp., Acute Immobilisation Test and reproduction Test. OECD Publishing, Paris. |
| [34] | OECD 201 (2011) Guidelines for the Testing of Chemicals, Section 2. No. 201. Freshwater Alga and Cyanobacteria, Growth Inhibition Test. OECD Publishing, Paris. |
| [35] | OECD 236 (2013) Guidelines for the Testing of Chemicals, Section 2. No. 236. Fish Embryo Acute Toxicity (FET) Test. OECD Publishing, Paris. |
| [36] |
Espiau F, Ortega J, Fernandez L, et al. (2011) Liquid-liquid equilibria in binary solutions formed by [Pyridinium-Derived][F4B] ionic liquids and alkanols: new experimental data and validation of a multiparametric model for correlating LLE data. Ind Eng Chem Res 50: 12259–12270. doi: 10.1021/ie201551w
|
| [37] | OECD 107 (1995) Guidelines for the Testing of Chemicals, Section 1. No. 107. Partition Coefficient (n-octanol/water): Shake Flask Method. OECD Publishing, Paris. |
| [38] |
Kim Y, Choi K, Jung J, et al. (2007) Aquatic toxicity of acetaminophen, carbamazepine, cimetidine, diltiazem and six major sulfonamides, and their potential ecological risks in Korea. Environ Int 33: 370–375. doi: 10.1016/j.envint.2006.11.017
|
| [39] |
Li MH (2013) Acute toxicity of 30 pharmaceutically active compounds to freshwater planarians, Dugesia japonica. Toxicol Environ Chem 95: 1157–1170. doi: 10.1080/02772248.2013.857671
|
| [40] |
Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ, et al. (2008) The occurrence of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs in surface water in South Wales, UK. Water Res 42: 3498–3518. doi: 10.1016/j.watres.2008.04.026
|
| [41] |
Passino DRM, Smith SB (1987) Acute bioassays and hazard evaluation of representative contaminants detected in great-lakes fish. Environ Toxicol Chem 6: 901–907. doi: 10.1002/etc.5620061111
|
| [42] |
Rocha R, Goncalves F, Marques C, et al. (2014) Environmental effects of anticholinesterasic therapeutic drugs on a crustacean species, Daphnia magna. Environ Sci Pollut R 21: 4418–4429. doi: 10.1007/s11356-013-2339-9
|
| [43] |
Parrella A, Lavorgna M, Criscuolo E, et al. (2014) Acute and chronic toxicity of six anticancer drugs on rotifers and crustaceans. Chemosphere 115: 59–66. doi: 10.1016/j.chemosphere.2014.01.013
|
| [44] |
Eguchi K, Nagase H, Ozawa M, et al. (2004) Evaluation of antimicrobial agents for veterinary use in the ecotoxicity test using microalgae. Chemosphere 57: 1733–1738. doi: 10.1016/j.chemosphere.2004.07.017
|
| [45] |
Elersek T, Milavec S, Korosec M, et al. (2016) Toxicity of the mixture of selected antineoplastic drugs against aquatic primary producers. Environ Sci Pollut R 23: 14780–14790. doi: 10.1007/s11356-015-6005-2
|
| [46] | Wagil M, Bialk-Bielinska A, Puckowski A, et al. (2014) Toxicity of anthelmintic drugs (fenbendazole and flubendazole) to aquatic organisms. Environ Sci Pollut Res Int 22: 2566–2573. |
| [47] |
Damasceno de Oliveira LL, Antunes SC, Goncalves F, et al. (2016) Acute and chronic ecotoxicological effects of four pharmaceuticals drugs on cladoceran Daphnia magna. Drug Chem Toxicol 39: 13–21. doi: 10.3109/01480545.2015.1029048
|
| [48] |
Li Q, Wang P, Chen L, et al. (2016) Acute toxicity and histopathological effects of naproxen in zebrafish (Danio rerio) early life stages. Environ Sci Pollut R 23: 18832–18841. doi: 10.1007/s11356-016-7092-4
|
| [49] |
Gheorghe S, Petre J, Lucaciu I, et al. (2016) Risk screening of pharmaceutical compounds in Romanian aquatic environment. Environ Monit Assess 188: 379. doi: 10.1007/s10661-016-5375-3
|
| [50] |
Brito RO, Marques EF, Silva SG, et al. (2009) Physicochemical and toxicological properties of novel amino acid-based amphiphiles and their spontaneously formed catanionic vesicles. Colloid Surface B 72: 80–87. doi: 10.1016/j.colsurfb.2009.03.017
|
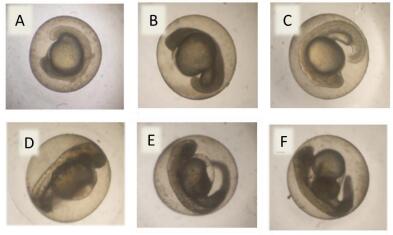
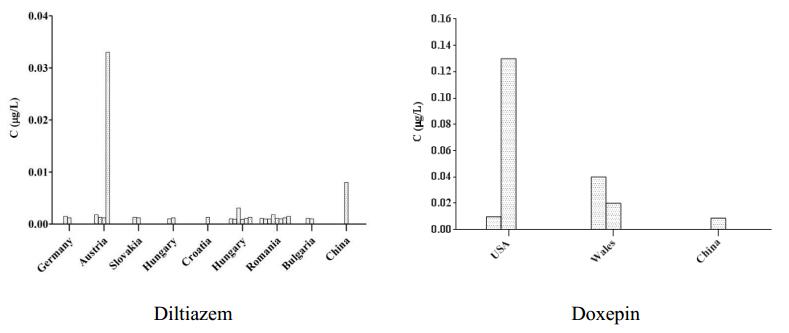
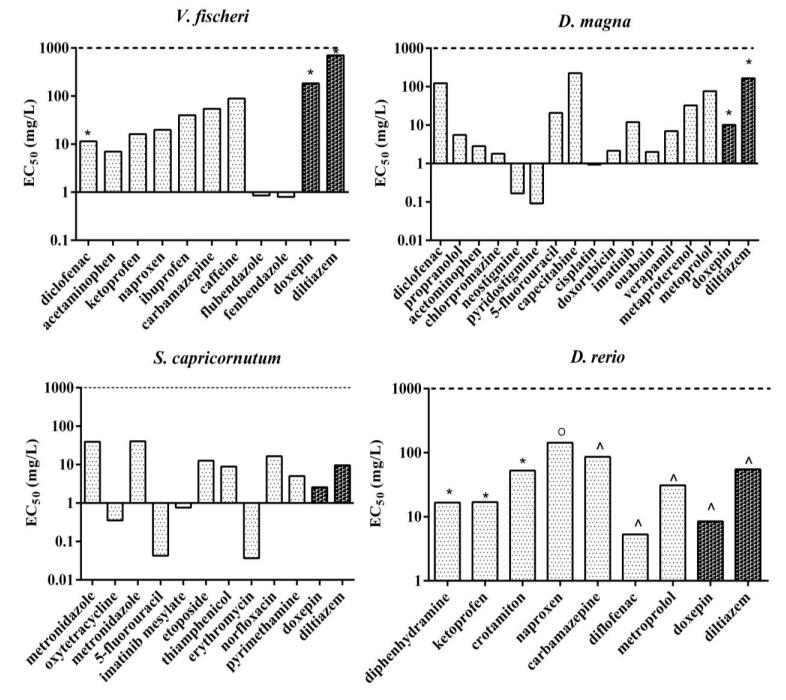
Figures(7) / Tables(3)

Natalia Ros, Laura Lomba, Ma Pilar Ribate, Estefania Zuriaga, Cristina B. García, Beatriz Giner. Acute lethal and sublethal effects of diltiazem and doxepin for four aquatic environmental bioindicators covering the trophic chain[J]. AIMS Environmental Science, 2018, 5(4): 229-243. doi: 10.3934/environsci.2018.4.229











 DownLoad:
DownLoad: