Citation: Milos Z. Markovic, Anne E. Perring, Ru-Shan Gao, Jin Liao, Andre Welti, Nick L. Wagner, Ilana B. Pollack, Ann M. Middlebrook, Thomas B. Ryerson, Michael K. Trainer, Carsten Warneke, Joost A. de Gouw, David W. Fahey, Philip Stier, Joshua P. Schwarz. Limited impact of sulfate-driven chemistry on black carbon aerosol aging in power plant plumes[J]. AIMS Environmental Science, 2018, 5(3): 195-215. doi: 10.3934/environsci.2018.3.195

| [1] |
Hansen J, Sato M, Ruedy R (1997) Radiative forcing and climate response. J Geophys Res 102: 6831–6864. doi: 10.1029/96JD03436

|
| [2] |
Jacobson MZ (2001) Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature 409: 695–697. doi: 10.1038/35055518
|
| [3] |
Ramanathan V, Carmichael G (2008) Global and regional climate changes due to black carbon. Nature Geosci 1: 221–227. doi: 10.1038/ngeo156
|
| [4] | IPCC (2013) Climate Change 2013 The Physical Science Basis-Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press. |
| [5] |
Ramanathan V, Crutzen PJ, Lelieveld J, et al. (2001) Indian Ocean Experiment: An integrated analysis of the climate forcing and effects of the great Indo-Asian haze. J Geophys Res 106: 28371–28398. doi: 10.1029/2001JD900133
|
| [6] | Tripathi SN, Dey S, Tare V, et al. (2005) Aerosol black carbon radiative forcing at an industrial city in northern India. Geophys Res Lett 32, L08802. |
| [7] |
Bond TC, Doherty SJ, Fahey DW, et al. (2013) Bounding the role of black carbon in the climate system: A scientific assessment. J Geophys Res-Atmos 118: 5380–5552. doi: 10.1002/jgrd.50171
|
| [8] |
Grieshop AP, Reynolds CCO, Kandlikar M, et al. (2009) A black-carbon mitigation wedge. Nature Geosci 2: 533–534. doi: 10.1038/ngeo595
|
| [9] |
Ramana MV, Ramanathan V, Feng Y, et al. (2010) Warming influenced by the ratio of black carbon to sulphate and the black-carbon source. Nature Geosci 3: 542–545. doi: 10.1038/ngeo918
|
| [10] |
Shindell D, Kuylenstierna JCI, Vignati E, et al. (2012) Simultaneously Mitigating Near-Term Climate Change and Improving Human Health and Food Security. Science 335: 183–189. doi: 10.1126/science.1210026
|
| [11] |
Nakicenovic N (2000) Greenhouse gas emissions scenarios. Technol Forecast Soc Change 65: 149–166. doi: 10.1016/S0040-1625(00)00094-9
|
| [12] |
Weingartner E, Burtscher H, Baltensperger U (1997) Hygroscopic properties of carbon and diesel soot particles. Atmos Environ 31: 2311–2327. doi: 10.1016/S1352-2310(97)00023-X
|
| [13] |
Schwarz JP, Perring AE, Markovic MZ, et al. (2015) Technique and theoretical approach for quantifying the hygroscopicity of black-carbon-containing aerosol using a single particle soot photometer. J Aerosol Sci 81: 110–126. doi: 10.1016/j.jaerosci.2014.11.009
|
| [14] | Stier P, Seinfeld JH, Kinne S, et al. (2006) Impact of nonabsorbing anthropogenic aerosols on clear-sky atmospheric absorption. J Geophys Res-Atmos 111, 2006JD007147. |
| [15] |
Subramanian R, Kok GL, Baumgardner D, et al. (2010) Black carbon over Mexico: the effect of atmospheric transport on mixing state, mass absorption cross-section, and BC/CO ratios. Atmos Chem Phys 10: 219–237. doi: 10.5194/acp-10-219-2010
|
| [16] | Chung SH, Seinfeld JH (2005) Climate response of direct radiative forcing of anthropogenic black carbon. J Geophys Res-Atmos 110. |
| [17] | Jacobson MZ (2002) Control of fossil-fuel particulate black carbon and organic matter, possibly the most effective method of slowing global warming. J Geophys Res-Atmos 107, 2001JD01376. |
| [18] | Lesins G, Chylek P, Lohmann U (2002) A study of internal and external mixing scenarios and its effect on aerosol optical properties and direct radiative forcing. J Geophys Res 107, 2001JD000973. |
| [19] |
Martins JV, Artaxo P, Liousse C, et al. (1998) Effects of black carbon content, particle size, and mixing on light absorption by aerosols from biomass burning in Brazil. J Geophys Res 103: 32041–32050. doi: 10.1029/98JD02593
|
| [20] |
Moffet RC, Prather KA (2009) In-situ measurements of the mixing state and optical properties of soot with implications for radiative forcing estimates. P Natl Acad SCI USA 106: 11872–11877. doi: 10.1073/pnas.0900040106
|
| [21] |
Lim S, Lee M, Kim SW, et al. (2018) Sulfate alters aerosol absorption properties in East Asian outflow. Nat Sci Rep 8: 5172. doi: 10.1038/s41598-018-23021-1
|
| [22] |
Wu Y, Cheng I, Zheng L, et al. (2016) Black carbon radiative forcing at TOA decreased during aging. Nat Sci Rep 6: 38592. doi: 10.1038/srep38592
|
| [23] |
Cooke WF, Liousse C, Cachier H, et al. (1999) Construction of a 1 degrees x 1 degrees fossil fuel emission data set for carbonaceous aerosol and implementation and radiative impact in the ECHAM4 model. J Geophys Res 104: 22137–22162. doi: 10.1029/1999JD900187
|
| [24] |
Croft B, Lohmann U, von Salzen K (2005) Black carbon ageing in the Canadian Centre for Climate modelling and analysis atmospheric general circulation model. Atmos Chem Phys 5: 1931–1949. doi: 10.5194/acp-5-1931-2005
|
| [25] |
Koch D (2001) Transport and direct radiative forcing of carbonaceous and sulfate aerosols in the GISS GCM. J Geophys Res 106: 20311–20332. doi: 10.1029/2001JD900038
|
| [26] |
Riemer N, Vogel H, Vogel B (2004) Soot aging time scales in polluted regions during day and night. Atmos Chem Phys 4: 1885–1893. doi: 10.5194/acp-4-1885-2004
|
| [27] |
Lund M, Berntsen K, Samset BH (2017) Sensitivity of black carbon concentrations and climate impact to aging and scavenging in OsloCTM2–M7. Atmos Chem Phys 17: 6003–6022. doi: 10.5194/acp-17-6003-2017
|
| [28] |
Bond TC, Bergstrom RW (2006) Light absorption by carbonaceous particles: An investigative review. Aerosol Sci Tech 40: 27–67. doi: 10.1080/02786820500421521
|
| [29] | Bond TC, Habib G, Bergstrom RW (2006) Limitations in the enhancement of visible light absorption due to mixing state. J Geophys Res-Atmos 111, 2006JD007315. |
| [30] | Cross ES, Onasch TB, Ahern A, et al. (2010b) Soot Particle Studies-Instrument Inter-Comparison-Project Overview. Aerosol Sci Tech 44: 592–611. |
| [31] |
Myhre G (2009) Consistency Between Satellite-Derived and Modeled Estimates of the Direct Aerosol Effect. Science 325: 187–190. doi: 10.1126/science.1174461
|
| [32] | Schnaiter M, Linke C, Mohler O, et al. (2005) Absorption amplification of black carbon internally mixed with secondary organic aerosol. J Geophys Res-Atmos 110, 2005JD006046. |
| [33] | Schwarz JP, Spackman JR, Fahey DW, et al. (2008) Coatings and their enhancement of black carbon light absorption in the tropical atmosphere. J Geophys Res 113, 2007JD009042. |
| [34] |
Lack DA, Langridge JM, Bahreini R, et al. (2012) Brown carbon and internal mixing in biomass burning particles. P Natl Acad Sci USA 109: 14802–14807. doi: 10.1073/pnas.1206575109
|
| [35] |
Wang Q, Huang RJ, Cao J, et al. (2014) Mixing state of black carbon aerosol in a heavily polluted urban area of China: Implications for light absorption enhancement. Aerosol Sci Tech 48: 689–697. doi: 10.1080/02786826.2014.917758
|
| [36] |
Cappa CD, Onasch TB, Massoli P, et al. (2012) Radiative Absorption Enhancements Due to the Mixing State of Atmospheric Black Carbon. Science 337: 1078–1081. doi: 10.1126/science.1223447
|
| [37] |
Lan ZJ, Huang XF, Yu KY, et al. (2013) Light absorption of black carbon aerosol and its enhancement by mixing state in an urban atmosphere in South China. Atmos Environ 69: 118–123. doi: 10.1016/j.atmosenv.2012.12.009
|
| [38] | Fierce L, Riemer N, Bond TC (2015) Explaining variance in black carbon's aging timescale. Atmos Chem Phys 15: 3173–3191. |
| [39] |
Warneke C, Trainer M, de Gouw, et al. (2016) Instrumentation and measurement strategy for the NOAA SENEX aircraft campaign as part of the Southeast Atmosphere Study 2013. Atmos Meas Tech 9: 3063–3093. doi: 10.5194/amt-9-3063-2016
|
| [40] | Attwood AR, Washenfelder RA, Brock CA, et al. (2014) Trends in sulfate and organic aerosol mass in the Southeast U.S.: Impact on aerosol optical depth and radiative forcing. Geophys Res Lett 41: 7701–7709. |
| [41] |
Brock CA, Wagner NL, Anderson BE, et al. (2015) Aerosol optical properties in the southeastern United States in summer–Part 2: Sensitivity of aerosol optical depth to relative humidity and aerosol parameters. Atmos Chem Phys Discuss 15: 31471–31499. doi: 10.5194/acpd-15-31471-2015
|
| [42] | Brock CA, Wagner NL, Anderson BE, et al. (2015b) Aerosol optical properties in the southeastern United States in summer–Part 1: Hygroscopic growth. Atmos Chem Phys Discuss 2015: 25695–25738. |
| [43] | Hand JL, Copeland SA, Day DE, et al. (2011) Spatial and Seasonal Patterns and Temporal Variability of Haze and its Constituents in the United States: Report V. Colorado State University, Fort Collins CO, 2011. |
| [44] |
Hidy GM, Blanchard CL, Baumann K, et al. (2014) Chemical climatology of the southeastern United States, 1999–2013. Atmos Chem Phys 14: 11893–11914. doi: 10.5194/acp-14-11893-2014
|
| [45] |
Kim PS, Jacob DJ, Fisher JA, et al. (2015) Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model. Atmos Chem Phys 15: 10411–10433. doi: 10.5194/acp-15-10411-2015
|
| [46] |
Wagner NL, Brock CA, Angevine WM, et al. (2015) In situ vertical profiles of aerosol extinction, mass, and composition over the southeast United States during SENEX and SEAC4RS: observations of a modest aerosol enhancement aloft. Atmos Chem Phys 15: 7085–7102. doi: 10.5194/acp-15-7085-2015
|
| [47] |
Washenfelder RA, Attwood AR, Brock CA, et al. (2015) Biomass burning dominates brown carbon absorption in the rural southeastern United States. Geophys Res Lett 42: 653–664. doi: 10.1002/2014GL062444
|
| [48] | Miller BG (2010) Clean Coal Engineering Technology. Elsevier Inc, Oxford, 375–481. |
| [49] | Zhang Y, Vijayaraghavan K, Wen XY, et al. (2009) Probing into regional ozone and particulate matter pollution in the United States: 1. A 1 year CMAQ simulation and evaluation using surface and satellite data. J Geophys Res-Atmos 114: 2009JD0011898. |
| [50] | Jacob DJ (1999) Introduction to Atmospheric Chemistry, Princeton University Press, Princeton, NJ. |
| [51] | Kulmala M, Kerminen VM (2008) On the formation and growth of atmospheric nanoparticles. Atmos Res 90: 32–150. |
| [52] |
Junkermann W, Vogel B, Sutton MA (2011) The climate penalty for clean fossil fuel combustion. Atmos Chem Phys 11: 12917–12924. doi: 10.5194/acp-11-12917-2011
|
| [53] | Junkermann W, Hagemann R, Vogel B (2011b) Nucleation in the Karlsruhe plume during the COPS/TRACKS-Lagrange experiment. Q J Roy Meteor Soc 137: 267–274. |
| [54] |
Lonsdale CR, Stevens RG, Brock CA, et al. (2012) The effect of coal-fired power-plant SO2 and NOx control technologies on aerosol nucleation in the source plumes. Atmos Chem Phys 12: 11519–11531. doi: 10.5194/acp-12-11519-2012
|
| [55] |
Petzold A, Ogren JA, Fiebig M, et al. (2013) Recommendations for reporting "black carbon" measurements. Atmos Chem Phys 13: 8365–8379. doi: 10.5194/acp-13-8365-2013
|
| [56] |
Schwarz JP, Spackman JR, Gao RS, et al. (2010) The Detection Efficiency of the Single Particle Soot Photometer. Aerosol Sci Tech 44: 612–628. doi: 10.1080/02786826.2010.481298
|
| [57] | Schwarz JP, Gao RS, Spackman JR, et al. (2008) Measurement of the mixing state, mass, and optical size of individual black carbon particles in urban and biomass burning emissions. Geophys Res Lett 35, L13810, 2008GL033968. |
| [58] | Cross ES, Onasch TB, Ahern A, et al. (2010a) Intercomparison study of black carbon measurements. Abstracts of Papers of the American Chemical Society, 1155 16TH ST, NW, WASHINGTON, DC 20036 USA: AMER CHEMICAL SOC, 240. |
| [59] | Schwarz JP, Gao RS, Fahey DW, et al. (2006) Single-particle measurements of midlatitude black carbon and light-scattering aerosols from the boundary layer to the lower stratosphere. J Geophys Res 111(D16), D16207, 2006JD007076. |
| [60] | Murphy DM, Cziczo DJ, Hudson PK, et al. (2004) Particle generation and resuspension in aircraft inlets when flying in clouds. Aerosol Sci Tech 38: 400–408. |
| [61] |
Lance S, Brock CA, Rogers D, et al. (2010) Water droplet calibration of the Cloud Droplet Probe (CDP) and in-flight performance in liquid, ice and mixed-phase clouds during ARCPAC. Atmos Meas Tech 3: 1683–1706. doi: 10.5194/amt-3-1683-2010
|
| [62] |
Baumgardner D, Popovicheva O, Allan J, et al. (2012) Soot reference materials for instrument calibration and intercomparisons: a workshop summary with recommendations. Atmos Meas Tech 5: 1869–1887. doi: 10.5194/amt-5-1869-2012
|
| [63] | Moteki N, Kondo Y (2010) Dependence of Laser-Induced Incandescence on Physical Properties of Black Carbon Aerosols: Measurements and Theoretical Interpretation. Aero Sci Technol 44: 663–675. |
| [64] |
Gysel M, Laborde M, Olfert JS, et al. (2011) Effective density of Aquadag and fullerene soot black carbon reference materials used for SP2 calibration. Atmos Meas Technol 4: 2851–2858. doi: 10.5194/amt-4-2851-2011
|
| [65] |
Gao RS, Schwarz JP, Kelly KK, et al. (2007) A novel method for estimating light-scattering properties of soot aerosols using a modified single-particle soot photometer. Aerosol Sci Tech 41: 125–135. doi: 10.1080/02786820601118398
|
| [66] |
Petters MD, Kreidenweis SM (2007) A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos Chem Phys 7: 1961–1971. doi: 10.5194/acp-7-1961-2007
|
| [67] | Bahreini R, Ervens B, Middlebrook AM, et al. (2009) Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas. J Geophys Res-Atmos 114, D00F16, 2008JD011493. |
| [68] |
Liao J, Brock CB, Murphy DM, et al. (2017) Single-particle measurements of bouncing particles and in situ collection efficiency from an airborne aerosol mass spectrometer (AMS) with light-scattering detection. Atmos Meas Tech 10: 3801–3820. doi: 10.5194/amt-10-3801-2017
|
| [69] |
Brock CA, Schroder F, Karcher B, et al. (2000) Ultrafine particle size distributions measured in aircraft exhaust plumes. J Geophys Res 105: 26555–26567. doi: 10.1029/2000JD900360
|
| [70] |
Brock C A, Cozic J, Bahreini R, et al. (2011) Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project. Atmos Chem Phys 11: 2423–2453. doi: 10.5194/acp-11-2423-2011
|
| [71] |
Pollack IB, Lerner BM, Ryerson TB (2010) Evaluation of ultraviolet light-emitting diodes for detection of atmospheric NO2 by photolysis-chemiluminescence. J Atmos Chem 65: 111–125. doi: 10.1007/s10874-011-9184-3
|
| [72] |
Ryerson TB, Williams EJ, Fehsenfeld FC (2000) An efficient photolysis system for fast-response NO2 measurements. J Geophys Res 105: 26447–26461. doi: 10.1029/2000JD900389
|
| [73] |
Ryerson TB, Huey LG, Knapp K, et al. (1999) Design and initial characterization of an inlet for gas-phase NOy measurements from aircraft. J Geophys Res 104: 5483–5492. doi: 10.1029/1998JD100087
|
| [74] |
Holloway JS, Jakoubek RO, Parrish DD, et al. (2000) Airborne intercomparison of vacuum ultraviolet fluorescence and tunable diode laser absorption measurements of tropospheric carbon monoxide. J Geophys Res 105: 24251–24261. doi: 10.1029/2000JD900237
|
| [75] | Jacobson MZ (2012) Investigating cloud absorption effects: Global absorption properties of black carbon, tar balls, and soil dust in clouds and aerosols. J Geophys Res-Atmos 117, 2011JD017218. |
| [76] |
Lack DA, Cappa CD (2010) Impact of brown and clear carbon on light absorption enhancement, single scatter albedo and absorption wavelength dependence of black carbon. Atmos Chem Phys 10: 4207–4220. doi: 10.5194/acp-10-4207-2010
|
| [77] |
Saleh R, Robinson ES, Tkacik DS, et al. (2014) Brownness of organics in aerosols from biomass burning linked to their black carbon content. Nature Geosci 7: 647–650. doi: 10.1038/ngeo2220
|
| [78] |
Qiu C, Khalizov AF, Zhang R (2012) Soot aging from OH-initiated oxidation of toluene. Environ Sci Technol 46: 9464–9472. doi: 10.1021/es301883y
|
| [79] |
China S, Scarnato B, Owen RC, et al. (2015) Morphology and mixing state of aged soot particles at a remote marine free troposphere site: Implications for optical properties. Geophys Res Lett 42: 1243–1250. doi: 10.1002/2014GL062404
|
| [80] | Reimer N, West M, Zaveri RA, et al. (2009) Simulating the evolution of soot mixing state with a particle-resolved aerosol model. J Geophys Res 114(D9), 2008JD011073. |
| [81] |
O'Donnel D, Tsigaridis K, Feichter J (2011) Estimating the direct and indirect effects of secondary organic aerosols using ECHAM5-HAM. Atmos Chem Phys 11: 8635–8559. doi: 10.5194/acp-11-8635-2011
|
| [82] |
Zhang K, O'Donnell D, Kazil J, et al. (2012) The global aerosol-climate model ECHAM-HAM, version 2: sensitivity to improvements in process representations. Atmos Chem Phys 12: 8911–8949. doi: 10.5194/acp-12-8911-2012
|
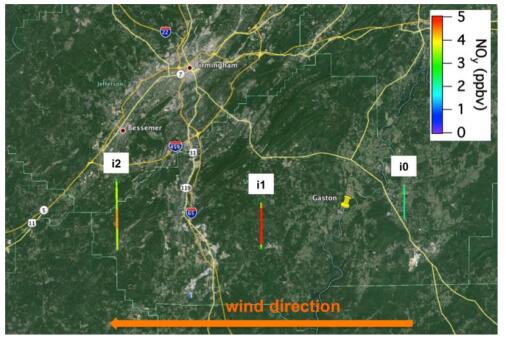
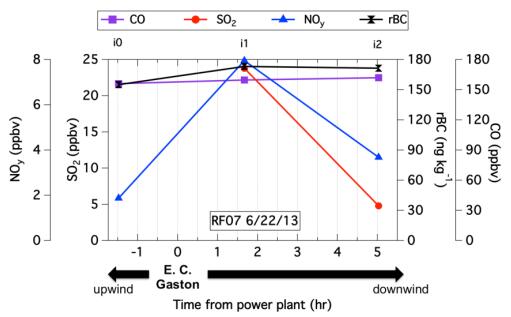
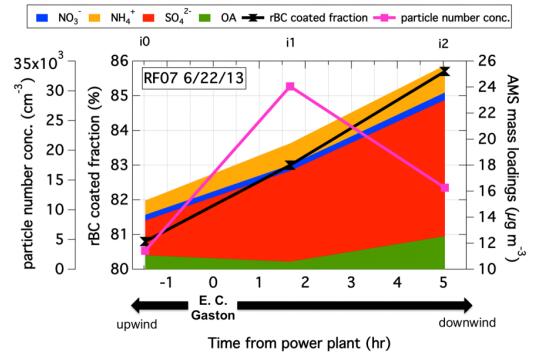
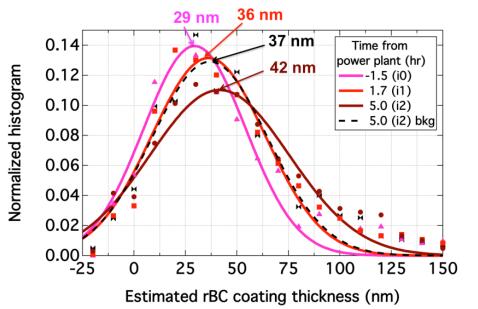
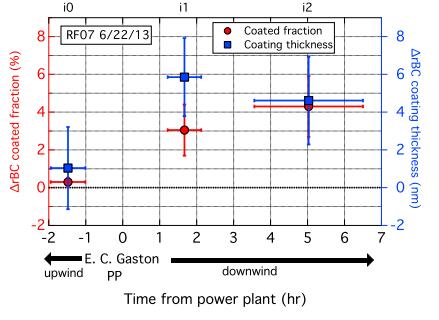
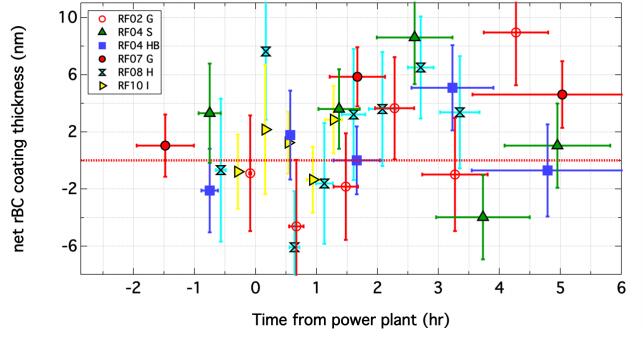
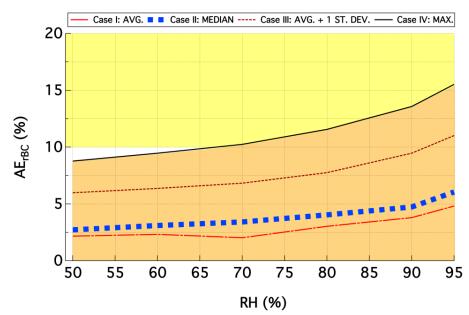
Figures(7) / Tables(1)

Milos Z. Markovic, Anne E. Perring, Ru-Shan Gao, Jin Liao, Andre Welti, Nick L. Wagner, Ilana B. Pollack, Ann M. Middlebrook, Thomas B. Ryerson, Michael K. Trainer, Carsten Warneke, Joost A. de Gouw, David W. Fahey, Philip Stier, Joshua P. Schwarz. Limited impact of sulfate-driven chemistry on black carbon aerosol aging in power plant plumes[J]. AIMS Environmental Science, 2018, 5(3): 195-215. doi: 10.3934/environsci.2018.3.195







 DownLoad:
DownLoad: