Citation: Wenyuan Xie, Jason Wei Jun Low, Arunmozhiarasi Armugam, Kandiah Jeyaseelan, Yen Wah Tong. Regulation of Aquaporin Z osmotic permeability in ABA tri-block copolymer[J]. AIMS Biophysics, 2015, 2(3): 381-397. doi: 10.3934/biophy.2015.3.381

| [1] |
Kumar M, Grzelakowski M, Zilles J, et al. (2007) Highly permeable polymeri membranes based on the incorporation of the functional water channel protein Aquaporin Z. PNAS 104: 20719-20724. doi: 10.1073/pnas.0708762104

|
| [2] | Service RF (2006) Desalination freshens up. Science 313:1088-1090. |
| [3] | Tal A (2006) Seeking sustainability: Israel's evolving water management strategy. Science 313: 1081-1084. |
| [4] |
Discher BM, Won YY, Ege DS, et al. (1999) Polymersomes: tough vesicles made from diblock copolymers. Science 284:1143-1146. doi: 10.1126/science.284.5417.1143
|
| [5] |
Calamita G, Kempf B, Rudd KE, et al. (1997) The aquaporin-Z water channel gene of Escherichia coli: Structure, organization and phylogeny. Biology of the Cell 89: 321-329. doi: 10.1111/j.1768-322X.1997.tb01029.x
|
| [6] |
Calamita G, Bishai WR, Preston GM, et al. (1995) Molecular cloning and characterization of AqpZ, a water channel from Escherichia coli. J Biol Chem 270: 29063-29066. doi: 10.1074/jbc.270.49.29063
|
| [7] | Scheuring S, Ringler P, Borgnia M, et al. (1999) High resultion AFM topographs of the Escherichia coli water channel aquaporin Z. EMBO J 18: 4981-4987. |
| [8] | Gorin MB, Yancey SB, Cline J, et al. (1984) The major intrinsic protein (MIP) of the bovine lens fiber membrane: Characterization and structure based on cDNA cloning. Cell 39: 49-59. |
| [9] |
Ishibashi K, Kuwahara M, Gu Y, et al. (1997) Cloning and functional expression of a new water channel abundantly expressed in the testis permeable to water, glycerol and urea. J Biol Chem 272: 20782-20786. doi: 10.1074/jbc.272.33.20782
|
| [10] | Soupene E, King N, Lee H, et al. (2001) Aquaporin Z of Escherichia coli: Reassessment of Its Regulation and Physiological Role. J Bacter 184: 4304-4307. |
| [11] |
Calamita G, Kempf B, Bonhivers B, et al. (1998) Regulation of the Escherichia coli water channel gene AqpZ. Proc Natl Acad Sci U S A 95: 3627-3631. doi: 10.1073/pnas.95.7.3627
|
| [12] | Borgnia MJ, Kozono D, Calamita G, et al. (1999) Funcation Reconstitution and Characterization of AqpZ, the E. coli Water Channel Protein. J Mol Biol 291: 1169-1179. |
| [13] |
Kozono D, Yasui M, King LS, et al. (2002). Aquaporin water channels: atomic structure molecular dynamics meet clinical medicine. J Clin Inves 109: 1395-1399. doi: 10.1172/JCI0215851
|
| [14] |
Nemeth-Cahalan KL, Hall JE (2000) pH and Calcium Regulate the Water Permeability of Aquaporin 0. J Biol Chem 275: 6777-6782. doi: 10.1074/jbc.275.10.6777
|
| [15] | Cahalan K, Kalman K, Hall JE (2004) Molecular Basis of pH and Ca2+ Regulation of Aquaporin Water Permeability. J Gen Physiol 123: 573-580 |
| [16] | Zhou W, Jones SW (1996) The effects of external pH on calcium channel currents in bullfrog sympathetic neurons. Biophys J 70: 1326-1334 |
| [17] | Gonen T, Walz T (2006) The structure of aquaporins. Q Rev Biophys 39: 361-396. |
| [18] |
Chaumont F, Moshelion F, Daniels MJ (2005) Regulation of plant aquaporin activity. Biol Cell 97: 749-764. doi: 10.1042/BC20040133
|
| [19] |
Tong J, Canty JT, Briggs MM, et al. (2013) The water permeability of lens aquaporin-0 depends on its lipid bilayer environment. Exp Eye Res 113: 32-40. doi: 10.1016/j.exer.2013.04.022
|
| [20] |
Andersen OS, Bruno MJ, Sun H, et al. (2007) Single-molecule methods for monitoring changes in bilayer elastic properties. Meth Mol Biol 400: 543-570 doi: 10.1007/978-1-59745-519-0_37
|
| [21] |
Hong H, Tamm LK (2004) Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci U S A 101: 4065-4070. doi: 10.1073/pnas.0400358101
|
| [22] |
Nyholm TK, Ozdirekcan S, Killian JA (2007) How protein transmembrane segments sense the lipid environment. Biochemistry 46: 1457-1465. doi: 10.1021/bi061941c
|
| [23] |
Phillips R, Ursell T, Wiggins P, et al. (2009) Emerging roles for lipids in shaping membrane-protein function. Nature 459: 379-385. doi: 10.1038/nature08147
|
| [24] | Yuan C, O'Connell RJ, Jacob RF, et al. (2007) Regulation of the gating of BKCa channel by lipid bilayer thickness. J Biol Chem 282: 7276-7286. |
| [25] |
Dumas F, Tocanne JF, Leblanc G, et al. (2000) Consequences of hydrophobic mismatch between lipids and melibiose permease on melibiose transport. Biochem 39: 4846-4854. doi: 10.1021/bi992634s
|
| [26] |
Perozo E, Kloda A, Cortes DM, et al. (2002) Physical principles underlying the transduction of bilayer deformation forces during mechano senditive channel gating. Nat Struct Biol 9: 696-703. doi: 10.1038/nsb827
|
| [27] |
Xie W, He F, Wang B, et al. (2013) An aquaporin-based vesicle-embedded polymeric membrane for low energy water filtration. J Mater Chem A 1: 7592-7600. doi: 10.1039/c3ta10731k
|
| [28] | Wang H, Chung TS, Tong YW, et al. (2011) Preparation and characterization of pore-suspending biomimetic membranes embedded with Aquaporin Z on carboxylated polyethylene glycol polymer cushion. Soft Matter 7: 7274-7280. |
| [29] | Wang H, Chung TS, Tong YW, et al. (2012) Highly permeable and selective pore-spanning biomimetic membrane embedded with aquaporin Z. Small 8: 1185-1190, 1125. |
| [30] | Duong PHH, Chung TS, Jeyaseelan K, et al. (2012) Planar biomimetic aquaporin-incorporated triblock copolymer membranes on porous alumina supports for nanofiltration. J Membr Sci 409: 34-43. |
| [31] | Zhong PS, Chung TS, Jeyaseelan K, et al. (2012) Aquaporin-embedded biomimetic membranes for nanofiltration. J Membr Sci 407: 27-33. |
| [32] | Savage DF, Egea PF, Colmenares YR, et al. (2013) Architecture and selectivity in aquaporins: 2.5A X-Ray Structure of Aquaporin Z. PLoS Biol 1: 334-340. |
| [33] |
Nielsen CH (2009) Biomimetic membranes for sensor and separation applications. Bioanal Chem 395: 697-718. doi: 10.1007/s00216-009-2960-0
|
| [34] | Discher DE, Eisenberg A(2002) Polymer vesicles. Science 297: 967-973. |
| [35] |
Ahmed F, Photos PJ, Discher DE (2006) Polymersomes as viral capsid mimics. Drug Develop Res 67: 4-14. doi: 10.1002/ddr.20062
|
| [36] |
Lewis BA, Engelman DM (1983) Lipid Bilayer Thickness Varies Linearly with Acyl Chain Length in Fluid Phosphatidylcholine Vesicles. J Mol Biol 166: 211-217. doi: 10.1016/S0022-2836(83)80007-2
|
| [37] |
Dave PC, Tiburu EK, Damodaran K, et al. (2004) Investigating Structural Changes in the Lipid Bilayer upon Insertion of the Transmembrane Domain of the Membrane-Bound Protein Phospholamban Utilizing 31P and 2H Solid-State NMR Spectroscopy. Biophys J 86: 1564-1573. doi: 10.1016/S0006-3495(04)74224-1
|
| [38] | Marsh D (2008) Energetics of Hydrophobic Matching in Lipid-Protein Interactions. Biophys J 94: 3996-4013. |
| [39] |
Xu Q, Kim M, David Ho KW, et al. (2008) Membrane Hydrocarbon Thickness Modulates the Dynamics of a Membrane Transport Protein. Biophys J 95: 2849-2858. doi: 10.1529/biophysj.108.133629
|
| [40] |
He F, Tong YW (2014) A mechanistic study on amphiphilic block co-polymer poly(butadiene-b-(ethylene oxide)) vesicles reveals the water permeation mechanism through a polymeric bilayer. RSC Adv 4: 15304-15313. doi: 10.1039/c3ra48063a
|
| [41] |
Yang B, Verkman AS (1997) Water and Glycerol Permeabilities of Aquaporins 1-5 and MIP Determined Quantitatively by Expression of Epitope-tagged Constructs in Xenopus Oocytes. J Biol Chem 272: 16140-16146. doi: 10.1074/jbc.272.26.16140
|
| [42] |
Mehdizadeh H, Dickson JM, Eriksson PK (1989) Temperature effects on the performance of thin-film composite, aromatic polyamide membranes. Ind Eng Chem Res 28: 814-824. doi: 10.1021/ie00090a025
|
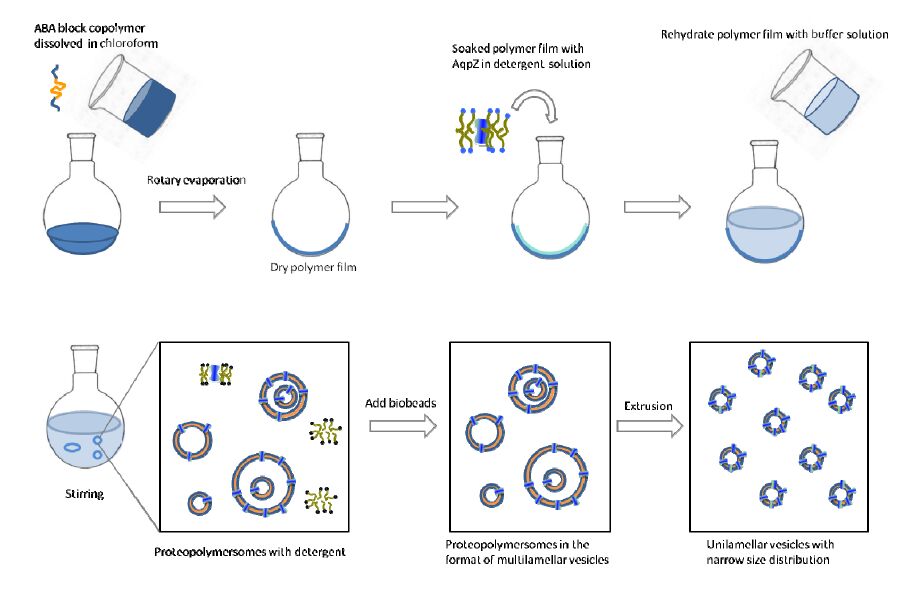
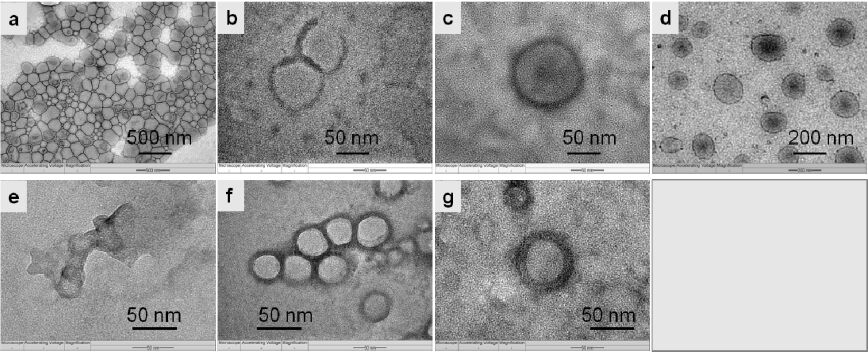
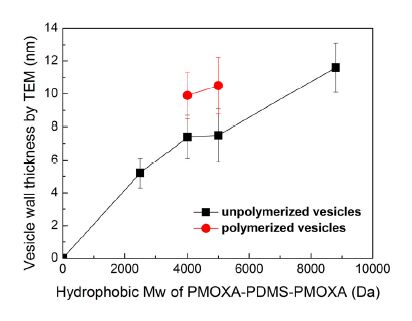
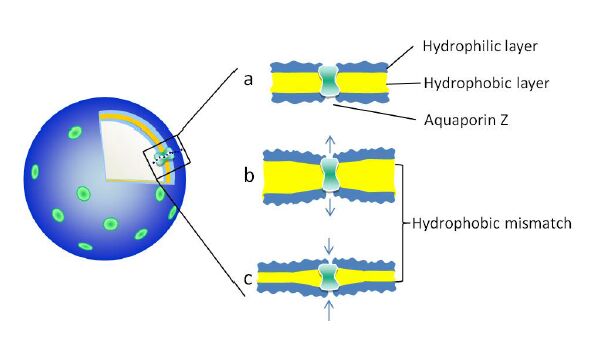
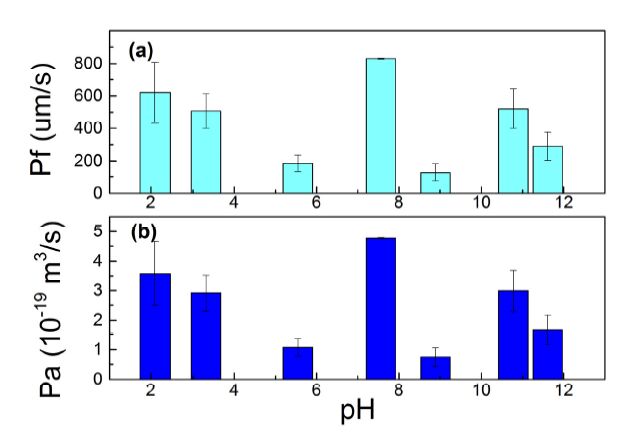
Figures(9) / Tables(1)

Wenyuan Xie, Jason Wei Jun Low, Arunmozhiarasi Armugam, Kandiah Jeyaseelan, Yen Wah Tong. Regulation of Aquaporin Z osmotic permeability in ABA tri-block copolymer[J]. AIMS Biophysics, 2015, 2(3): 381-397. doi: 10.3934/biophy.2015.3.381







 DownLoad:
DownLoad: