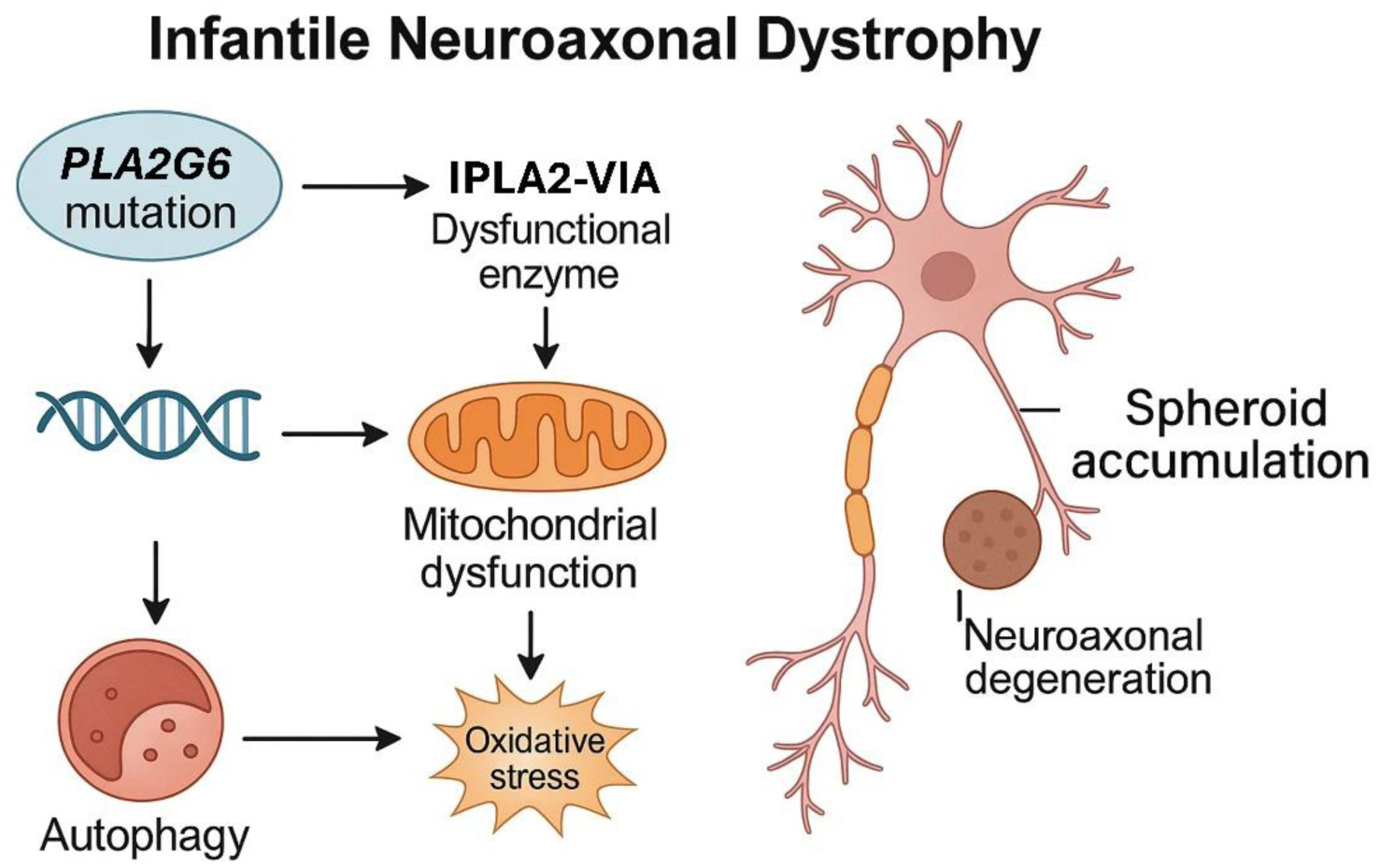
Infantile neuroaxonal dystrophy (INAD), also known as PLA2G6-associated neurodegeneration (PLAN), is a rare, early-onset, autosomal recessively inherited neurodegenerative disease belonging to the group of neurodegenerations with brain iron accumulation (NBIA). The main cause of this disease is bi-allelic mutations in the PLA2G6 gene, which codes for the enzyme phospholipase A2 type VI. Clinically, it manifests with progressive neurodevelopmental impairment, psychomotor regression, movement disorders, and pyramidal signs. Initially described in the 1950s, the classical form presents in the first two years of life, although later-onset variants are recognized. At the neuropathological level, INAD is characterized by the presence of neuroaxonal spheroids, which are dilations of degenerated axons, located mainly in the white matter, basal ganglia, and cerebellum. INAD is considered a rare or ultra-rare disease, with an estimated prevalence of approximately 1 per million individuals. Diagnosis requires a comprehensive evaluation combining clinical with neuroimaging studies, mainly magnetic resonance imaging (MRI), and genetic analysis. MRI may reveal early cerebellar atrophy and a low-intensity signal in the globus pallidus on iron-sensitive sequences, indicative of iron accumulation. Currently, there is no curative treatment for INAD, so management focuses on providing palliative care and symptom control using a multidisciplinary approach. However, various therapeutic strategies are being investigated, including gene therapy to correct the genetic defect, as well as approaches to modulate pathological pathways such as lipid peroxidation and iron accumulation.
Citation: María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos. Infantile neuroaxonal dystrophy: Molecular mechanisms and pathogenesis of PLA2G6-associated neurodegeneration[J]. AIMS Neuroscience, 2025, 12(2): 180-202. doi: 10.3934/Neuroscience.2025011
Infantile neuroaxonal dystrophy (INAD), also known as PLA2G6-associated neurodegeneration (PLAN), is a rare, early-onset, autosomal recessively inherited neurodegenerative disease belonging to the group of neurodegenerations with brain iron accumulation (NBIA). The main cause of this disease is bi-allelic mutations in the PLA2G6 gene, which codes for the enzyme phospholipase A2 type VI. Clinically, it manifests with progressive neurodevelopmental impairment, psychomotor regression, movement disorders, and pyramidal signs. Initially described in the 1950s, the classical form presents in the first two years of life, although later-onset variants are recognized. At the neuropathological level, INAD is characterized by the presence of neuroaxonal spheroids, which are dilations of degenerated axons, located mainly in the white matter, basal ganglia, and cerebellum. INAD is considered a rare or ultra-rare disease, with an estimated prevalence of approximately 1 per million individuals. Diagnosis requires a comprehensive evaluation combining clinical with neuroimaging studies, mainly magnetic resonance imaging (MRI), and genetic analysis. MRI may reveal early cerebellar atrophy and a low-intensity signal in the globus pallidus on iron-sensitive sequences, indicative of iron accumulation. Currently, there is no curative treatment for INAD, so management focuses on providing palliative care and symptom control using a multidisciplinary approach. However, various therapeutic strategies are being investigated, including gene therapy to correct the genetic defect, as well as approaches to modulate pathological pathways such as lipid peroxidation and iron accumulation.

| [1] |
Altuame FD, Foskett G, Atwal PS, et al. (2020) The natural history of infantile neuroaxonal dystrophy. Orphanet J Rare Dis 15: 109. https://doi.org/10.1186/s13023-020-01355-2 
|
| [2] |
Babin PL, Rao SNR, Chacko A, et al. (2018) Infantile Neuroaxonal Dystrophy: Diagnosis and Possible Treatments. Front Genet 9: 597. https://doi.org/10.3389/fgene.2018.00597
|
| [3] |
Fatima A, Abuhijleh SA, Fatah A, et al. (2024) Infantile Neuroaxonal Dystrophy: Case Report and Review of Literature. Medicina (Kaunas) 60: 1322. https://doi.org/10.3390/medicina60081322
|
| [4] |
Lyu Y, Wang T, Lin M, et al. (2024) A rare inherited homozygous missense variant in PLA2G6 influences susceptibility to infantile neuroaxonal dystrophy: a case report. Transl Pediatr 13: 484-491. https://doi.org/10.21037/tp-23-568
|
| [5] | Adams D, Midei M, Dastgir J, et al. (2020) Treatment of infantile neuroaxonal dystrophy with RT001: A di-deuterated ethyl ester of linoleic acid: Report of two cases. JIMD Rep 54: 54-60. https://doi.org/10.1002/jmd2.12116 |
| [6] | Ansari B, Nasiri J, Namazi H, et al. (2022) Infantile Neuroaxonal Dystrophy in Two Cases: Siblings with Different Presentations. Iran J Child Neurol 16: 193-198. https://doi.org/10.22037/ijcn.v16i2.30864 |
| [7] |
Iodice A, Spagnoli C, Salerno GG, et al. (2017) Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: An update for the diagnosis. Brain Dev 39: 93-100. https://doi.org/10.1016/j.braindev.2016.08.012
|
| [8] |
Al-Maawali A, Yoon G, Feigenbaum AS, et al. (2016) Validation of the finding of hypertrophy the clava in infantile neuroaxonal dystrophy/PLA2G6 by biometric analysis. Neuroradiology 58: 1035-1042. https://doi.org/10.1007/s00234-016-1726-6
|
| [9] |
Atwal PS, Midei M, Adams D, et al. (2020) The infantile neuroaxonal dystrophy rating scale (INAD-RS). Orphanet J Rare Dis 15: 195. https://doi.org/10.1186/s13023-020-01479-5
|
| [10] |
Cheema AN, Shi R, Kamboh MI (2025) Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy. Int J Mol Sci 26: 352. https://doi.org/10.3390/ijms26010352
|
| [11] |
Elsayed LEO, Mohammed IN, Hamed AAA, et al. (2018) Case report of a novel homozygous splice site mutation in PLA2G6 gene causing infantile neuroaxonal dystrophy in a Sudanese family. BMC Med Genet 19: 72. https://doi.org/10.1186/s12881-018-0592-y
|
| [12] |
Fukusumi H, Togo K, Beck G, et al. (2023) Human induced pluripotent stem cell line (ONHi001-A) generated from a patient with infantile neuroaxonal dystrophy having PLA2G6 c.517C > T (p.Q173X) and c.1634A > G (p.K545R) compound heterozygous mutations. Stem Cell Res 69: 103122. https://doi.org/10.1016/j.scr.2023.103122
|
| [13] | Hao Y, Chen D, Zhang G, et al. (2020) Successful clinical application of pre-implantation genetic diagnosis for infantile neuroaxonal dystrophy. Exp Ther Med 19: 956-964. https://doi.org/10.3892/etm.2019.8302 |
| [14] |
Iannello G, Graziano C, Cenacchi G, et al. (2017) A new PLA2G6 mutation in a family with infantile neuroaxonal dystrophy. J Neurol Sci 381: 209-212. https://doi.org/10.1016/j.jns.2017.08.3260
|
| [15] |
Li H, Zou Y, Bao X, et al. (2016) Monozygotic twins with infantile neuroaxonal dystrophy: A case report and literature review. Exp Ther Med 12: 3387-3389. https://doi.org/10.3892/etm.2016.3761
|
| [16] |
Li L, Fong CY, Tay CG, et al. (2020) Infantile neuroaxonal dystrophy in a pair of Malaysian siblings with progressive cerebellar atrophy: Description of an expanded phenotype with novel PLA2G6 variants. J Clin Neurosci 71: 289-292. https://doi.org/10.1016/j.jocn.2019.08.111
|
| [17] |
Rostampour D, Zolfaghari MR, Gholami M (2022) Novel insertion mutation in the PLA2G6 gene in an Iranian family with infantile neuroaxonal dystrophy. J Clin Lab Anal 36: e24253. https://doi.org/10.1002/jcla.24253
|
| [18] | Wang B, Wu D, Tang J (2018) Infantile neuroaxonal dystrophy caused by PLA2G6 gene mutation in a Chinese patient: A case report. Exp Ther Med 16: 1290-1294. https://doi.org/10.3892/etm.2018.6347 |
| [19] |
Yamamoto T, Shimojima K, Shibata T, et al. (2015) Novel PLA2G6 mutations associated with an exonic deletion due to non-allelic homologous recombination in a patient with infantile neuroaxonal dystrophy. Hum Genome Var 2: 15048. https://doi.org/10.1038/hgv.2015.48
|
| [20] |
Zou Y, Luo H, Yuan H, et al. (2022) Identification of a Novel Nonsense Mutation in PLA2G6 and Prenatal Diagnosis in a Chinese Family With Infantile Neuroaxonal Dystrophy. Front Neurol 13: 904027. https://doi.org/10.3389/fneur.2022.904027
|
| [21] |
Kapoor S, Shah MH, Singh N, et al. (2016) Genetic Analysis of PLA2G6 in 22 Indian Families with Infantile Neuroaxonal Dystrophy, Atypical Late-Onset Neuroaxonal Dystrophy and Dystonia Parkinsonism Complex. PLoS One 11: e0155605. https://doi.org/10.1371/journal.pone.0155605
|
| [22] |
Gregory A, Westaway SK, Holm IE, et al. (2008) Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 71: 1402-1409. https://doi.org/10.1212/01.wnl.0000327094.67726.28
|
| [23] |
Carrilho I, Santos M, Guimaraes A, et al. (2008) Infantile neuroaxonal dystrophy: what's most important for the diagnosis?. Eur J Paediatr Neurol 12: 491-500. https://doi.org/10.1016/j.ejpn.2008.01.005
|
| [24] |
Wilson JL, Soo AKS, Gregory A, et al. (2025) Consensus Clinical Management Guideline for PLA2G6-Associated Neurodegeneration (PLAN). J Child Neurol 40: 8830738251323649. https://doi.org/10.1177/08830738251323649
|
| [25] |
Sadeh M (2009) Neurodegeneration associated with genetic defects in phospholipase A2. Neurology 73: 819. https://doi.org/10.1212/WNL.0b013e3181b2851b
|
| [26] |
Wu Y, Jiang Y, Gao Z, et al. (2009) Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur J Neurol 16: 240-245. https://doi.org/10.1111/j.1468-1331.2008.02397.x
|
| [27] |
Guo YP, Tang BS, Guo JF (2018) PLA2G6-Associated Neurodegeneration (PLAN): Review of Clinical Phenotypes and Genotypes. Front Neurol 9: 1100. https://doi.org/10.3389/fneur.2018.01100
|
| [28] | Lu Y, Liu CH, Wang Y (2019) [Clinical features of infantile neuroaxonal dystrophy and PLA2G6 gene testing]. Zhongguo Dang Dai Er Ke Za Zhi 21: 851-855. https://doi.org/10.7499/j.issn.1008-8830.2019.09.002 |
| [29] |
Strokin M, Reiser G (2017) Neurons and astrocytes in an infantile neuroaxonal dystrophy (INAD) mouse model show characteristic alterations in glutamate-induced Ca(2+) signaling. Neurochem Int 108: 121-132. https://doi.org/10.1016/j.neuint.2017.03.004
|
| [30] |
Strokin M, Reiser G (2016) Mitochondria from a mouse model of the human infantile neuroaxonal dystrophy (INAD) with genetic defects in VIA iPLA2 have disturbed Ca(2+) regulation with reduction in Ca(2+) capacity. Neurochem Int 99: 187-193. https://doi.org/10.1016/j.neuint.2016.07.002
|
| [31] |
Kurian MA, McNeill A, Lin JP, et al. (2011) Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol 53: 394-404. https://doi.org/10.1111/j.1469-8749.2011.03955.x
|
| [32] |
Hogarth P, Gregory A, Kruer MC, et al. (2013) New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 80: 268-275. https://doi.org/10.1212/WNL.0b013e31827e07be
|
| [33] |
Sumi-Akamaru H, Beck G, Kato S, et al. (2015) Neuroaxonal dystrophy in PLA2G6 knockout mice. Neuropathology 35: 289-302. https://doi.org/10.1111/neup.12202
|
| [34] |
Wada H, Yasuda T, Miura I, et al. (2009) Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in Pla2g6. Am J Pathol 175: 2257-2263. https://doi.org/10.2353/ajpath.2009.090343
|
| [35] |
Zhao Z, Wang J, Zhao C, et al. (2011) Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. PLoS One 6: e26991. https://doi.org/10.1371/journal.pone.0026991
|
| [36] |
Beck G, Sugiura Y, Shinzawa K, et al. (2011) Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 31: 11411-11420. https://doi.org/10.1523/JNEUROSCI.0345-11.2011
|
| [37] |
Kasinathan A, Ahuja CK, Singhi P (2018) Spastic paraparesis with basal ganglia changes: Infantile neuroaxonal dystrophy. Neurol India 66: 264. https://doi.org/10.4103/0028-3886.222887
|
| [38] |
Kinghorn KJ, Castillo-Quan JI, Li L, et al. (2016) Reply: Glial mitochondropathy in infantile neuroaxonal dystrophy: pathophysiological and therapeutic implications. Brain 139: e68. https://doi.org/10.1093/brain/aww185
|
| [39] |
Catania A, Battini R, Pippucci T, et al. (2018) R106C TFG variant causes infantile neuroaxonal dystrophy “plus” syndrome. Neurogenetics 19: 179-187. https://doi.org/10.1007/s10048-018-0552-x
|
| [40] |
Farrar MA, Teoh HL, Brammah S, et al. (2016) Glial mitochondropathy in infantile neuroaxonal dystrophy: pathophysiological and therapeutic implications. Brain 139: e67. https://doi.org/10.1093/brain/aww174
|
| [41] | Fujiwara T, Watanabe Y, Tanaka H, et al. (2019) Quantitative susceptibility mapping (QSM) evaluation of infantile neuroaxonal dystrophy. BJR Case Rep 5: 20180078. https://doi.org/10.1259/bjrcr.20180078 |
| [42] |
Morgan NV, Westaway SK, Morton JE, et al. (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38: 752-754. https://doi.org/10.1038/ng1826
|
| [43] |
Frattini D, Nardocci N, Pascarella R, et al. (2015) Downbeat nystagmus as the presenting symptom of infantile neuroaxonal dystrophy: a case report. Brain Dev 37: 270-272. https://doi.org/10.1016/j.braindev.2014.04.010
|
| [44] |
Singh S, Mishra SC, Israrahmed A, et al. (2021) Typical MRI features of PLA2G6 mutation-related phospholipase-associated neurodegeneration (PLAN)/infantile neuroaxonal dystrophy (INAD). BMJ Case Rep 14: e242586. https://doi.org/10.1136/bcr-2021-242586
|
| [45] |
Mascalchi M, Mari F, Berti B, et al. (2017) Fast Progression of Cerebellar Atrophy in PLA2G6-Associated Infantile Neuronal Axonal Dystrophy. Cerebellum 16: 742-745. https://doi.org/10.1007/s12311-017-0843-z
|
| [46] |
Zhong Q, Zhong Q, Lai B (2024) Infantile neuroaxonal dystrophy causing iron deposition in the bilateral globus pallidus. QJM 117: 137-138. https://doi.org/10.1093/qjmed/hcad217
|
| [47] |
Riku Y, Ikeuchi T, Yoshino H, et al. (2013) Extensive aggregation of alpha-synuclein and tau in juvenile-onset neuroaxonal dystrophy: an autopsied individual with a novel mutation in the PLA2G6 gene-splicing site. Acta Neuropathol Commun 1: 12. https://doi.org/10.1186/2051-5960-1-12
|
| [48] |
Santucci M, Ambrosetto G, Scaduto MC, et al. (2001) Ictal and nonictal paroxysmal events in infantile neuroaxonal dystrophy: polygraphic study of a case. Epilepsia 42: 1074-1077. https://doi.org/10.1046/j.1528-1157.2001.0420081074.x
|
| [49] |
Sinskey JL, Holzman RS (2017) Perioperative considerations in infantile neuroaxonal dystrophy. Paediatr Anaesth 27: 322-324. https://doi.org/10.1111/pan.13081
|
| [50] |
Fusco C, Frattini D, Panteghini C, et al. (2015) A case of infantile neuroaxonal dystrophy of neonatal onset. J Child Neurol 30: 368-370. https://doi.org/10.1177/0883073814535493
|
| [51] | Gong WD, Tao G, Zhao TT, et al. (2023) Diagnosis, treatment and genetic analysis of a child with infantile neuroaxonal dystrophy. Yi Chuan 45: 617-623. https://doi.org/10.16288/j.yczz.23-034 |
| [52] |
Gregory A, Polster BJ, Hayflick SJ (2009) Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet 46: 73-80. https://doi.org/10.1136/jmg.2008.061929
|
| [53] | Wang J, Wu W, Chen X, et al. (2016) [A novel homozygous mutation in PLA2G6 gene causes infantile neuroaxonal dystrophy in a case]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 33: 64-67. https://doi.org/10.3760/cma.j.issn.1003-9406.2016.01.016 |
| [54] |
Cheng YC, Lin HI, Syu SH, et al. (2019) Reprogramming of a human induced pluripotent stem cell (iPSC) line (IBMSi012-A) from an early-onset Parkinson's disease patient harboring a homozygous p.D331Y mutation in the PLA2G6 gene. Stem Cell Res 37: 101432. https://doi.org/10.1016/j.scr.2019.101432
|
| [55] |
Chiu CC, Wang HL, Weng YH, et al. (2019) Generation of induced pluripotent stem cells from a young-onset Parkinson's disease patient carrying the compound heterozygous PLA2G6 p.D331Y/p.M358IfsX mutations. Stem Cell Res 40: 101552. https://doi.org/10.1016/j.scr.2019.101552
|
| [56] |
Gopurappilly R (2021) Pluripotent Stem Cell Derived Neurons as In Vitro Models for Studying Autosomal Recessive Parkinson's Disease (ARPD): PLA2G6 and Other Gene Loci. Adv Exp Med Biol 1347: 115-133. https://doi.org/10.1007/5584_2021_643
|
| [57] |
Huang J, Jiang Q, Pang D, et al. (2024) Generation of induced pluripotent stem cell line LNDWCHi001-A from a patient with early-onset Parkinson's disease carrying the homozygous c.1898C > T (p. A633V) mutation in the PLA2G6 gene. Stem Cell Res 75: 103305. https://doi.org/10.1016/j.scr.2024.103305
|
| [58] |
Lyamzaev KG, Huan H, Panteleeva AA, et al. (2024) Exogenous Iron Induces Mitochondrial Lipid Peroxidation, Lipofuscin Accumulation, and Ferroptosis in H9c2 Cardiomyocytes. Biomolecules 14: 730. https://doi.org/10.3390/biom14060730
|
| [59] | Tan J, Yan T, Chang R, et al. (2020) [Analysis of PLA2G6 gene variant in a family affected with infantile neuroaxonal dystrophy]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 37: 21-24. https://doi.org/10.3760/cma.j.issn.1003-9406.2020.01.006 |
Figures(1) / Tables(2)

María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos. Infantile neuroaxonal dystrophy: Molecular mechanisms and pathogenesis of PLA2G6-associated neurodegeneration[J]. AIMS Neuroscience, 2025, 12(2): 180-202. doi: 10.3934/Neuroscience.2025011







 DownLoad:
DownLoad: