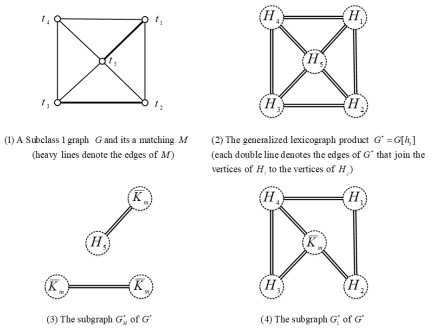
An edge-coloring of a graph $ G $ is an assignment of colors to its edges so that no two edges incident to the same vertex receive the same color. The chromatic index of $ G $, denoted by $ \chi'(G) $, is the least $ k $ for which $ G $ has a $ k $ edge-coloring. Graphs with $ \chi'(G) = \Delta(G) $ are said to be Class 1, and graphs with $ \chi'(G) = \Delta(G)+1 $ are said to be Class 2. Let $ G $ be a graph with $ V(G) = \{t_1, t_2, \ldots, t_n\} $, $ n\geq2 $, and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ be a sequence of vertex-disjoint graphs with $ V(H_i) = \{(t_i, y_1), (t_i, y_2), \ldots, (t_i, y_{m_i})\} $, $ m_i\geq1 $. The generalized lexicographic product $ G[h_n] $ of $ G $ and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ is a simple graph with vertex set $ \bigcup_{i = 1}^{n}V(H_i) $, in which $ (t_i, y_p) $ is adjacent to $ (t_j, y_q) $ if and only if either $ t_i = t_j $ and $ (t_i, y_p)(t_i, y_q)\in E(H_i) $ or $ t_it_j\in E(G) $. If $ G $ is a complete graph with order 2, then $ G[h_2] $ denotes a join $ H_1+H_2 $ of vertex-disjoint graphs $ H_1 $ and $ H_2 $. If $ H_i\cong H $ for $ i = 1, 2, \ldots, n $, then $ G[h_n] = G[H] $, where $ G[H] $ denotes the lexicographic product of two graphs $ G $ and $ H $. In this paper, we provide sufficient conditions for the generalized lexicographic product $ G[h_n] $ of $ G $ and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ to be Class 1, where all graphs in $ h_n $ have the same number of vertices.
Citation: Shuangliang Tian, Ping Chen. Edge-coloring of generalized lexicographic product of graphs[J]. AIMS Mathematics, 2024, 9(6): 15988-15995. doi: 10.3934/math.2024774
An edge-coloring of a graph $ G $ is an assignment of colors to its edges so that no two edges incident to the same vertex receive the same color. The chromatic index of $ G $, denoted by $ \chi'(G) $, is the least $ k $ for which $ G $ has a $ k $ edge-coloring. Graphs with $ \chi'(G) = \Delta(G) $ are said to be Class 1, and graphs with $ \chi'(G) = \Delta(G)+1 $ are said to be Class 2. Let $ G $ be a graph with $ V(G) = \{t_1, t_2, \ldots, t_n\} $, $ n\geq2 $, and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ be a sequence of vertex-disjoint graphs with $ V(H_i) = \{(t_i, y_1), (t_i, y_2), \ldots, (t_i, y_{m_i})\} $, $ m_i\geq1 $. The generalized lexicographic product $ G[h_n] $ of $ G $ and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ is a simple graph with vertex set $ \bigcup_{i = 1}^{n}V(H_i) $, in which $ (t_i, y_p) $ is adjacent to $ (t_j, y_q) $ if and only if either $ t_i = t_j $ and $ (t_i, y_p)(t_i, y_q)\in E(H_i) $ or $ t_it_j\in E(G) $. If $ G $ is a complete graph with order 2, then $ G[h_2] $ denotes a join $ H_1+H_2 $ of vertex-disjoint graphs $ H_1 $ and $ H_2 $. If $ H_i\cong H $ for $ i = 1, 2, \ldots, n $, then $ G[h_n] = G[H] $, where $ G[H] $ denotes the lexicographic product of two graphs $ G $ and $ H $. In this paper, we provide sufficient conditions for the generalized lexicographic product $ G[h_n] $ of $ G $ and $ h_n = (H_i)_{i\in\{1, 2, \ldots, n\}} $ to be Class 1, where all graphs in $ h_n $ have the same number of vertices.

| [1] |
A. G. Chetwynd, A. J. W. Hilton, Regular graphs of high degree are 1-factorizable, Proc. London Math. Soc., 50 (1985), 193–206. https://doi.org/10.1112/plms/s3-50.2.193 doi: 10.1112/plms/s3-50.2.193

|
| [2] | R. Diestel, Graph Theory, Berlin: Springer-Verlag, 2017. |
| [3] |
I. Holyer, The NP-completeness of edge-coloring, SIAM J. Comput., 10 (1981), 718–720. https://doi.org/10.1137/0210055 doi: 10.1137/0210055
|
| [4] |
M. M. M. Jaradat, On the Anderson-Lipman conjecture and some related problems, Discrete Math., 297 (2005), 167–173. https://doi.org/10.1016/j.disc.2004.09.012 doi: 10.1016/j.disc.2004.09.012
|
| [5] | B. Mohar, On edge colorability of products of graphs, Publ. Inst. Math., 36 (1984), 13–16. |
| [6] |
C. De Simonea, A. Galluccio, Edge-colouring of joins of regular graphs Ⅰ, J. Comb. Optim., 18 (2009), 417–428. https://doi.org/10.1007/s10878-009-9235-8 doi: 10.1007/s10878-009-9235-8
|
| [7] |
C. De Simonea, A. Galluccio, Edge-colouring of joins of regular graphs Ⅱ, J. Comb. Optim., 25 (2013), 78–90. https://doi.org/10.1007/s10878-011-9420-4 doi: 10.1007/s10878-011-9420-4
|
| [8] |
C. De Simone, A. Galluccio, Edge-colouring of regular graphs of large degree, Theoret. Comput. Sci., 389 (2007), 91–99. https://doi.org/10.1016/j.tcs.2007.07.046 doi: 10.1016/j.tcs.2007.07.046
|
| [9] |
C. De Simonea, C. Picinin de Mello, Edge-colouring of join graphs, Theoret. Comput. Sci., 355 (2006), 364–370. https://doi.org/10.1016/j.tcs.2005.12.010 doi: 10.1016/j.tcs.2005.12.010
|
| [10] |
W. Szumny, I. Włoch, A. Włoch, On the existence and on the number of $(k, 1)$-kernels in the lexicographic product of graphs, Discrete Math., 308 (2008), 4616–4624. https://doi.org/10.1016/j.disc.2007.08.078 doi: 10.1016/j.disc.2007.08.078
|
| [11] | D. B. West, Introduction to Graph Theory, 2 Eds., Englewood Cliffs: Prentice-Hall, 2000. |

Figures(2)

Shuangliang Tian, Ping Chen. Edge-coloring of generalized lexicographic product of graphs[J]. AIMS Mathematics, 2024, 9(6): 15988-15995. doi: 10.3934/math.2024774







 DownLoad:
DownLoad: