Citation: Maurizio Garrione. Beams with an intermediate pier: Spectral properties, asymmetry and stability[J]. Mathematics in Engineering, 2021, 3(2): 1-21. doi: 10.3934/mine.2021016

| [1] | Burdina VI (1953) Boundedness of solutions of a system of differential equations. Dokl Akad Nauk SSSR 92: 603-606. |
| [2] | Cazenave T, Weissler FB (1996) Unstable simple modes of the nonlinear string. Quart Appl Math 54: 287-305. |
| [3] | Garrione M, Gazzola F (2017) Loss of energy concentration in nonlinear evolution beam equations. J Nonlinear Sci 27: 1789-1827. |
| [4] | Garrione M, Gazzola F (2019) Nonlinear Equations for Beams and Degenerate Plates with Piers, Cham: SpringerBriefs in Applied Sciences and Technology, Springer. |
| [5] | Garrione M, Gazzola F (2020) Linear theory for beams with intermediate piers. Commun Contemp Math DOI: 10.1142/S0219199719500810. |
| [6] | Gasparetto C, Gazzola F (2018) Resonance tongues for the Hill equation with Duffing coefficients and instabilities in a nonlinear beam equation. Commun Contemp Math 20: 1-22. |
| [7] | Gazzola F (2015) Mathematical Models for Suspension Bridges, Cham: Springer. |
| [8] | Gesztesy F, Weikard R (1996) Picard potentials and Hill's equation on a torus. Acta Math 176: 73-107. |
| [9] | Goldberg W, Hochstadt H (1996) On a Hill's equation with two gaps in its spectrum. SIAM J Math Anal 10: 1069-1076. |
| [10] | Harazaki I, Suzuki S, Okukawa A (2000) Suspension Bridges, Bridge Engineering Handbook, Boca Raton: CRC Press. |
| [11] | Holubová G, Nečesal P (2010) The Fučik spectra for multi-point boundary-value problems. Electronic H Diff Eq Conf 18: 33-44. |
| [12] | Yakubovich VA, Starzhinskii VM (1975) Linear Differential Equations with Periodic Coefficients, New York: J. Wiley & Sons. |
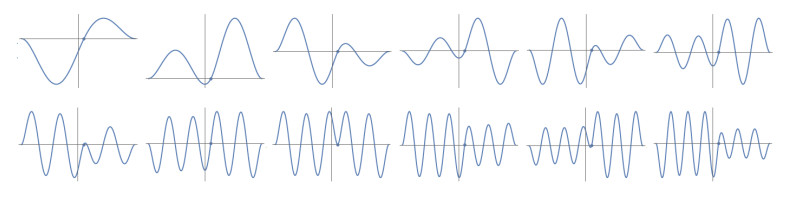
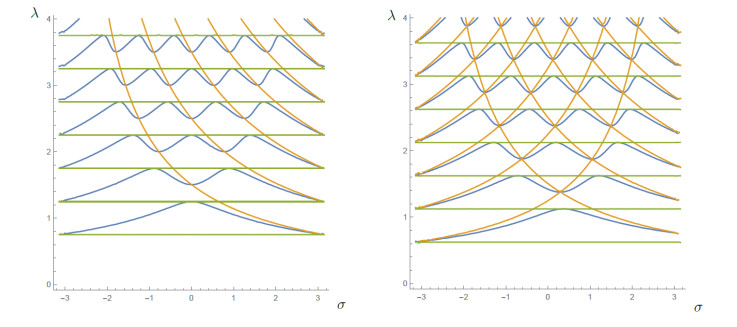
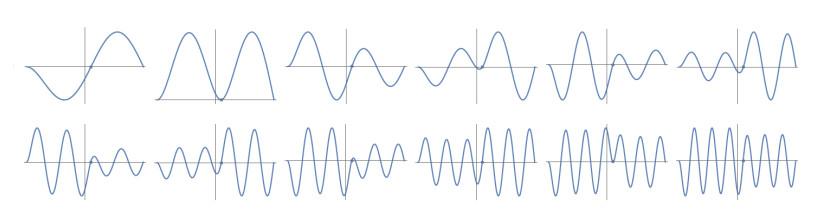
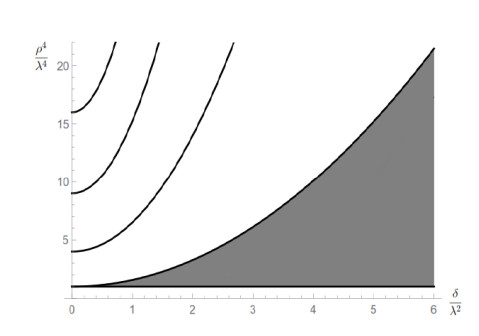
Figures(10) / Tables(1)

Maurizio Garrione. Beams with an intermediate pier: Spectral properties, asymmetry and stability[J]. Mathematics in Engineering, 2021, 3(2): 1-21. doi: 10.3934/mine.2021016







 DownLoad:
DownLoad: