Citation: Didier Pinault. N-Methyl D-Aspartate Receptor Antagonists Amplify Network Baseline Gamma Frequency (30–80 Hz) Oscillations: Noise and Signal[J]. AIMS Neuroscience, 2014, 1(2): 169-182. doi: 10.3934/Neuroscience.2014.2.169

| [1] |
Ben-Simon E, Podlipsky I, Arieli A, et al. (2008) Never resting brain simultaneous representation of two alpha related processes in humans. PLoS One 3: e3984. doi: 10.1371/journal.pone.0003984

|
| [2] |
de Munck JC, Goncalves SI, Huijboom L, et al. (2007) The hemodynamic response of the alpha rhythm an EEG/fMRI study. Neuroimage 35: 1142-1151. doi: 10.1016/j.neuroimage.2007.01.022
|
| [3] | Moss RA, Moss J. (2014) The role of dynamic columns in explaining gamma-band synchronization and NMDA receptors in cognitive functions. AIMS Neurosci 1: 65-88. |
| [4] | Cadonic C, Albensi BC. (2014) Oscillations and NMDA receptors their interplay create memories. AIMS Neurosci 1: 52-64. |
| [5] | Pinotsis D, Friston K. (2014) Gamma oscillations and neural field DCMs can reveal cortical excitability and microstructure. AIMS Neurosci 1: 18-38. |
| [6] |
Jasper HH. (1936) Cortical excitatory state and variability in human brain rhythms. Science 83:259-260. doi: 10.1126/science.83.2150.259
|
| [7] | Sheer DE. (1975) Behavior and brain electrical activity. New York and London: Plenum Press. |
| [8] | Sheer DE. (1989) Sensory and cognitive 40-Hz event-related potentials behavioral correlates, brain function and clinical application Brain Dynamics. Berlin: Springer, pp 339-374. |
| [9] |
Kulli J, Koch C. (1991) Does anesthesia cause loss of consciousness? Trends Neurosci 14: 6-10. doi: 10.1016/0166-2236(91)90172-Q
|
| [10] |
Ferri R, Cosentino FI, Elia M, et al. (2001) Relationship between Delta, Sigma, Beta, and Gamma EEG bands at REM sleep onset and REM sleep end. Clin Neurophysiol 112: 2046-2052. doi: 10.1016/S1388-2457(01)00656-3
|
| [11] |
Cantero JL, Atienza M, Madsen JR, et al. (2004) Gamma EEG dynamics in neocortex and hippocampus during human wakefulness and sleep. Neuroimage 22: 1271-1280. doi: 10.1016/j.neuroimage.2004.03.014
|
| [12] | Baldeweg T, Spence S, Hirsch SR, et al. (1998) Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet 352: 620-621. |
| [13] |
Becker C, Gramann K, Muller HJ, et al. (2009) Electrophysiological correlates of flicker-induced color hallucinations. Conscious Cogn 18: 266-276. doi: 10.1016/j.concog.2008.05.001
|
| [14] |
Behrendt RP. (2003) Hallucinations synchronisation of thalamocortical gamma oscillations underconstrained by sensory input. Conscious Cogn 12: 413-451. doi: 10.1016/S1053-8100(03)00017-5
|
| [15] |
Ffytche DH. (2008) The hodology of hallucinations. Cortex 44: 1067-1083. doi: 10.1016/j.cortex.2008.04.005
|
| [16] |
Spencer KM, Nestor PG, Perlmutter R, et al. (2004) Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci U S A 101: 17288-17293. doi: 10.1073/pnas.0406074101
|
| [17] |
Bartha R, Williamson PC, Drost DJ, et al. (1997) Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatr 54: 959-965. doi: 10.1001/archpsyc.1997.01830220085012
|
| [18] | Theberge J, Bartha R, Drost DJ, et al. (2002) Glutamate and glutamine measured with 4. 0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatr 159:1944-1946. |
| [19] |
Lutz A, Greischar LL, Rawlings NB, et al. (2004) Long-term meditators self-induce high-amplitude gamma synchrony during mental practice. Proc Natl Acad Sci USA 101: 16369-16373. doi: 10.1073/pnas.0407401101
|
| [20] | Joliot M, Ribary U, Llinas R. (1994) Human oscillatory brain activity near 40 Hz coexists with cognitive temporal binding. Proc Natl Acad Sci U S A 91: 11748-11751. |
| [21] |
Tallon-Baudry C, Bertrand O. (1999) Oscillatory gamma activity in humans and its role in object representation. Trends Cogn Sci 3: 151-162. doi: 10.1016/S1364-6613(99)01299-1
|
| [22] |
Varela F, Lachaux JP, Rodriguez E, et al. (2001) The brainweb phase synchronization and large-scale integration. Nat Rev Neurosci 2: 229-239. doi: 10.1038/35067550
|
| [23] | Zhang ZG, Hu L, Hung YS, et al. (2012) Gamma-band oscillations in the primary somatosensory cortex, a direct and obligatory correlate of subjective pain intensity. J Neurosci 32: 7429-7438. |
| [24] |
Buzsaki G, Chrobak JJ. (1995) Temporal structure in spatially organized neuronal ensembles a role for interneuronal networks. Curr Opin Neurobiol 5: 504-510. doi: 10.1016/0959-4388(95)80012-3
|
| [25] | Buzsaki G. (2006) Rhythms of the brain. Oxford University Press. |
| [26] |
Engel AK, Roelfsema PR, Fries P, Brecht M, Singer W. (1997) Role of the temporal domain for response selection and perceptual binding. Cereb Cortex 7: 571-582. doi: 10.1093/cercor/7.6.571
|
| [27] |
Fries P. (2009) Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu Rev Neurosci 32: 209-224. doi: 10.1146/annurev.neuro.051508.135603
|
| [28] |
Gray CM, Konig P, Engel AK, et al. (1989) Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature 338: 334-337. doi: 10.1038/338334a0
|
| [29] |
Singer W. (1999) Time as coding space? Curr Opin Neurobiol 9: 189-194. doi: 10.1016/S0959-4388(99)80026-9
|
| [30] |
Mantini D, Perrucci MG, Del GC, et al. (2007) Electrophysiological signatures of resting state networks in the human brain. Proc Natl Acad Sci U S A 104: 13170-13175. doi: 10.1073/pnas.0700668104
|
| [31] |
Buzsaki G, Draguhn A. (2004) Neuronal oscillations in cortical networks. Science 304: 1926-1929. doi: 10.1126/science.1099745
|
| [32] |
Buzsaki G, Wang XJ. (2012) Mechanisms of gamma oscillations. Annu Rev Neurosci 35: 203-225. doi: 10.1146/annurev-neuro-062111-150444
|
| [33] |
Steriade M. (2006) Grouping of brain rhythms in corticothalamic systems. Neurosci 137: 1087-1106. doi: 10.1016/j.neuroscience.2005.10.029
|
| [34] |
Roux F, Uhlhaas PJ. (2014) Working memory and neural oscillations alpha-gamma versus theta-gamma codes for distinct WM information? Trends Cogn Sci 18: 16-25. doi: 10.1016/j.tics.2013.10.010
|
| [35] |
Uhlhaas PJ, Roux F, Singer W, et al. (2009) The development of neural synchrony reflects late maturation and restructuring of functional networks in humans. Proc Natl Acad Sci U S A 106:9866-9871. doi: 10.1073/pnas.0900390106
|
| [36] | Woolsey TA, Van Der Loos H. (1970) The structural organization of layer IV in the somatosensory region. (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res 17: 205-242. |
| [37] |
Van Der Loos H. (1976) Barreloids in mouse somatosensory thalamus. Neurosci Lett 2: 1-6. doi: 10.1016/0304-3940(76)90036-7
|
| [38] |
Yang JW, An S, Sun JJ, et al. (2013) Thalamic network oscillations synchronize ontogenetic columns in the newborn rat barrel cortex. Cereb Cortex 23: 1299-1316. doi: 10.1093/cercor/bhs103
|
| [39] |
Minlebaev M, Colonnese M, Tsintsadze T, et al. (2011) Early gamma oscillations synchronize developing thalamus and cortex. Science 334: 226-229. doi: 10.1126/science.1210574
|
| [40] |
Pinault D, Deschenes M. (1992) Voltage-dependent 40-Hz oscillations in rat reticular thalamic neurons in vivo. Neurosci 51: 245-258. doi: 10.1016/0306-4522(92)90312-P
|
| [41] |
Pinault D. (2004) The thalamic reticular nucleus structure, function and concept. Brain Res Rev 46:1-31. doi: 10.1016/j.brainresrev.2004.04.008
|
| [42] | Mountcastle VB. (1957) Modality and topographic properties of single neurons of cat's somatic sensory cortex. J Neurophysiol 20: 408-434. |
| [43] | Mountcastle VB. (1997) The columnar organization of the neocortex. Brain 120. ( Pt 4): 701-722. |
| [44] |
Feldmeyer D, Brecht M, Helmchen F, et al. (2013) Barrel cortex function. Prog Neurobiol 103: 3-27. doi: 10.1016/j.pneurobio.2012.11.002
|
| [45] |
Horton JC, Adams DL. (2005) The cortical column a structure without a function. Philos Trans R Soc Lond B Biol Sci 360: 837-862. doi: 10.1098/rstb.2005.1623
|
| [46] |
Adler CM, Goldberg TE, Malhotra AK, et al. (1998) Effects of Ketamine on Thought Disorder, Working Memory, and Semantic Memory in Healthy Volunteers. Biological Psychiatr 43: 811-816. doi: 10.1016/S0006-3223(97)00556-8
|
| [47] |
Hetem LA, Danion JM, Diemunsch P, et al. (2000) Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology. (Berl) 152: 283-288. doi: 10.1007/s002130000511
|
| [48] | Krystal JH, Karper LP, Seibyl JP, et al. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatr 51: 199-214. |
| [49] |
Newcomer JW, Farber NB, Jevtovic-Todorovic V, et al. (1999) Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20:106-118. doi: 10.1016/S0893-133X(98)00067-0
|
| [50] | Fond G, Loundou A, Rabu C, et al. (2014) Ketamine administration in depressive disorders a systematic review and meta-analysis. Psychopharmacology. (Berl). In press. |
| [51] | McGirr A, Berlim MT, Bond DJ, et al. (2014) A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med 1-12. |
| [52] |
Zarate CA, Jr. , Singh JB, Carlson PJ, et al. (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatr 63: 856-864. doi: 10.1001/archpsyc.63.8.856
|
| [53] | Anticevic A, Corlett PR, Cole MW, et al. (2014) NMDA Receptor Antagonist Effects on Prefrontal Cortical Connectivity Better Model Early Than Chronic Schizophrenia. Biol Psychiatr [Epub ahead of print]. |
| [54] |
Driesen NR, McCarthy G, Bhagwagar Z, et al. (2013) Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatr 18: 1199-1204. doi: 10.1038/mp.2012.194
|
| [55] |
Hong LE, Summerfelt A, Buchanan RW, et al. (2010) Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 35:632-640. doi: 10.1038/npp.2009.168
|
| [56] | Pinault D. (2008) N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatr 63: 730-735. |
| [57] |
Chrobak JJ, Hinman JR, Sabolek HR. (2008) Revealing past memories proactive interference and ketamine-induced memory deficits. J Neurosci 28: 4512-4520. doi: 10.1523/JNEUROSCI.0742-07.2008
|
| [58] |
Kocsis B. (2012) Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatr 71: 987-995. doi: 10.1016/j.biopsych.2011.10.002
|
| [59] |
Ma J, Leung LS. (2007) The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology (Berl) 191: 961-974. doi: 10.1007/s00213-006-0667-x
|
| [60] |
Hakami T, Jones NC, Tolmacheva EA, et al. (2009) NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS One 4: e6755. doi: 10.1371/journal.pone.0006755
|
| [61] |
Ehrlichman RS, Gandal MJ, Maxwell CR, et al. (2009) N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience 158: 705-712. doi: 10.1016/j.neuroscience.2008.10.031
|
| [62] |
Hunt MJ, Raynaud B, Garcia R. (2006) Ketamine dose-dependently induces high-frequency oscillations in the nucleus accumbens in freely moving rats. Biol Psychiatr 60: 1206-1214. doi: 10.1016/j.biopsych.2006.01.020
|
| [63] |
Kulikova SP, Tolmacheva EA, Anderson P, Gaudias J, Adams BE, Zheng T, et al. (2012) Opposite effects of ketamine and deep brain stimulation on rat thalamocortical information processing. Eur J Neurosci 36: 3407-3419. doi: 10.1111/j.1460-9568.2012.08263.x
|
| [64] |
Molina LA, Skelin I, Gruber AJ. (2014) Acute NMDA receptor antagonism disrupts synchronization of action potential firing in rat prefrontal cortex. PLoS One 9: e85842. doi: 10.1371/journal.pone.0085842
|
| [65] |
Homayoun H, Moghaddam B. (2007) NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27: 11496-11500. doi: 10.1523/JNEUROSCI.2213-07.2007
|
| [66] |
Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, et al. (2000) Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex 10:1078-1092. doi: 10.1093/cercor/10.11.1078
|
| [67] |
Corlett PR, Honey GD, Fletcher PC. (2007) From prediction error to psychosis ketamine as a pharmacological model of delusions. J Psychopharmacol 21: 238-252. doi: 10.1177/0269881107077716
|
| [68] |
Adell A, Jimenez-Sanchez L, Lopez-Gil X, et al. (2012) Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr Bull 38: 9-14. doi: 10.1093/schbul/sbr133
|
| [69] |
Frohlich J, Van Horn JD. (2014) Reviewing the ketamine model for schizophrenia. J Psychopharmacol 28: 287-302. doi: 10.1177/0269881113512909
|
| [70] |
Gunduz-Bruce H. (2009) The acute effects of NMDA antagonism from the rodent to the human brain. Brain Res Rev 60: 279-286. doi: 10.1016/j.brainresrev.2008.07.006
|
| [71] |
Canolty RT, Knight RT. (2010) The functional role of cross-frequency coupling. Trends Cogn Sci 14:506-515. doi: 10.1016/j.tics.2010.09.001
|
| [72] |
Kirihara K, Rissling AJ, Swerdlow NR, et al. (2012) Hierarchical organization of gamma and theta oscillatory dynamics in schizophrenia. Biol Psychiatr 71: 873-880. doi: 10.1016/j.biopsych.2012.01.016
|
| [73] |
Jensen O, Colgin LL. (2007) Cross-frequency coupling between neuronal oscillations. Trends Cogn Sci 11: 267-269. doi: 10.1016/j.tics.2007.05.003
|
| [74] |
Lisman JE, Jensen O. (2013) The theta-gamma neural code. Neuron 77: 1002-1016. doi: 10.1016/j.neuron.2013.03.007
|
| [75] |
Palenicek T, Fujakova M, Brunovsky M, et al. (2011) Electroencephalographic spectral and coherence analysis of ketamine in rats correlation with behavioral effects and pharmacokinetics. Neuropsychobiology 63: 202-218. doi: 10.1159/000321803
|
| [76] |
Tsuda N, Hayashi K, Hagihira S, et al. (2007) Ketamine, an NMDA-antagonist, increases the oscillatory frequencies of alpha-peaks on the electroencephalographic power spectrum. Acta Anaesthesiol Scand 51: 472-481. doi: 10.1111/j.1399-6576.2006.01246.x
|
| [77] | Caixeta FV, Cornelio AM, Scheffer-Teixeira R, et al. (2013) Ketamine alters oscillatory coupling in the hippocampus. Sci Rep 3: 2348. |
| [78] |
Hiyoshi T, Kambe D, Karasawa J, et al. (2014) Differential effects of NMDA receptor antagonists at lower and higher doses on basal gamma band oscillation power in rat cortical electroencephalograms. Neuropharmacology 85: 384-396. doi: 10.1016/j.neuropharm.2014.05.037
|
| [79] |
Nicolas MJ, Lopez-Azcarate J, Valencia M, et al. (2011) Ketamine-induced oscillations in the motor circuit of the rat basal ganglia. PLoS One 6: e21814. doi: 10.1371/journal.pone.0021814
|
| [80] |
Buzsaki G. (1991) The thalamic clock emergent network properties. Neurosci 41: 351-364. doi: 10.1016/0306-4522(91)90332-I
|
| [81] | Friston KJ. (2002) Dysfunctional connectivity in schizophrenia. World Psychiatr 1: 66-71. |
| [82] | Melillo R, Leisman G. (2009) Autistic spectrum disorders as functional disconnection syndrome. Rev Neurosci 20: 111-131. |
| [83] |
de Haan W. , Pijnenburg YA, Strijers RL, et al. (2009) Functional neural network analysis in frontotemporal dementia and Alzheimer's disease using EEG and graph theory. BMC Neurosci 10:101. doi: 10.1186/1471-2202-10-101
|
| [84] |
Bokde AL, Ewers M, Hampel H. (2009) Assessing neuronal networks understanding Alzheimer's disease. Prog Neurobiol 89: 125-133. doi: 10.1016/j.pneurobio.2009.06.004
|
| [85] |
Popescu BO, Toescu EC, Popescu LM, et al. (2009) Blood-brain barrier alterations in ageing and dementia. J Neurol Sci 283: 99-106. doi: 10.1016/j.jns.2009.02.321
|
| [86] |
Herrmann CS, Demiralp T. (2005) Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol 116: 2719-2733. doi: 10.1016/j.clinph.2005.07.007
|
| [87] |
van Deursen JA, Vuurman EF, Verhey FR, et al. (2008) Increased EEG gamma band activity in Alzheimer's disease and mild cognitive impairment. J Neural Transm 115: 1301-1311. doi: 10.1007/s00702-008-0083-y
|
| [88] |
Yordanova J, Banaschewski T, Kolev V, et al. (2001) Abnormal early stages of task stimulus processing in children with attention-deficit hyperactivity disorder--evidence from event-related gamma oscillations. Clin Neurophysiol 112: 1096-1108. doi: 10.1016/S1388-2457(01)00524-7
|
| [89] | Spencer KM, Nestor PG, Niznikiewicz MA, et al. (2003) Abnormal neural synchrony in schizophrenia. J Neurosci 23: 7407-7411. |
| [90] |
Uhlhaas PJ, Singer W. (2006) Neural synchrony in brain disorders relevance for cognitive dysfunctions and pathophysiology. Neuron 52: 155-168. doi: 10.1016/j.neuron.2006.09.020
|
| [91] |
Whittington MA. (2008) Can brain rhythms inform on underlying pathology in schizophrenia? Biol Psychiatr 63: 728-729. doi: 10.1016/j.biopsych.2008.02.007
|
| [92] | Cronenwett WJ, Csernansky J. (2010) Thalamic pathology in schizophrenia. Curr Top Behav Neurosci 4: 509-528. |
| [93] |
Ferrarelli F, Peterson MJ, Sarasso S, et al. (2010) Thalamic dysfunction in schizophrenia suggested by whole-night deficits in slow and fast spindles. Am J Psychiatr 167: 1339-1348. doi: 10.1176/appi.ajp.2010.09121731
|
| [94] |
Lisman JE, Pi HJ, Zhang Y, et al. (2010) A thalamo-hippocampal-ventral tegmental area loop may produce the positive feedback that underlies the psychotic break in schizophrenia. Biol Psychiatr 68:17-24. doi: 10.1016/j.biopsych.2010.04.007
|
| [95] |
Pinault D. (2011) Dysfunctional thalamus-related networks in schizophrenia. Schizophr Bull 37:238-243. doi: 10.1093/schbul/sbq165
|
| [96] |
Watis L, Chen SH, Chua HC, et al. (2008) Glutamatergic abnormalities of the thalamus in schizophrenia a systematic review. J Neural Transm 115: 493-511. doi: 10.1007/s00702-007-0859-5
|
| [97] |
Zhang Y, Su TP, Liu B, et al. (2014) Disrupted thalamo-cortical connectivity in schizophrenia a morphometric correlation analysis. Schizophr Res 153: 129-135. doi: 10.1016/j.schres.2014.01.023
|
| [98] |
Javitt DC. (2007) Glutamate and schizophrenia phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol 78: 69-108. doi: 10.1016/S0074-7742(06)78003-5
|
| [99] |
Moghaddam B. (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40: 881-884. doi: 10.1016/S0896-6273(03)00757-8
|
| [100] |
Gandal MJ, Edgar JC, Klook K, et al. (2012) Gamma synchrony towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology 62: 1504-1518. doi: 10.1016/j.neuropharm.2011.02.007
|
| [101] |
Rolls ET, Loh M, Deco G, et al. (2008) Computational models of schizophrenia and dopamine modulation in the prefrontal cortex. Nat Rev Neurosci 9: 696-709. doi: 10.1038/nrn2462
|
| [102] |
Winterer G, Ziller M, Dorn H, et al. (2000) Schizophrenia reduced signal-to-noise ratio and impaired phase-locking during information processing. Clin Neurophysiol 111: 837-849. doi: 10.1016/S1388-2457(99)00322-3
|
| [103] |
Llinas RR, Ribary U, Jeanmonod D, et al. (1999) Thalamocortical dysrhythmia A neurological and neuropsychiatric syndrome characterized by magnetoencephalography. Proc Natl Acad Sci USA 96:15222-15227. doi: 10.1073/pnas.96.26.15222
|
| [104] |
Gonzalez-Burgos G, Lewis DA. (2008) GABA neurons and the mechanisms of network oscillations implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull 34: 944-961. doi: 10.1093/schbul/sbn070
|
| [105] |
Roopun AK, Cunningham MO, Racca C, et al. (2008) Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophr Bull 34: 962-973. doi: 10.1093/schbul/sbn059
|
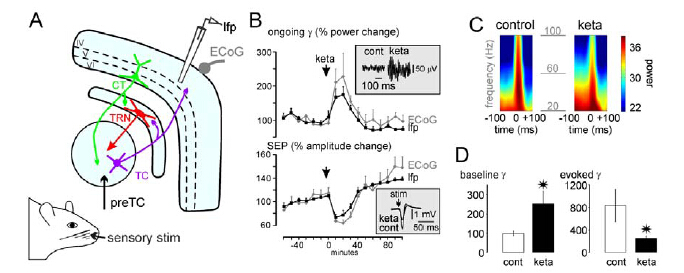
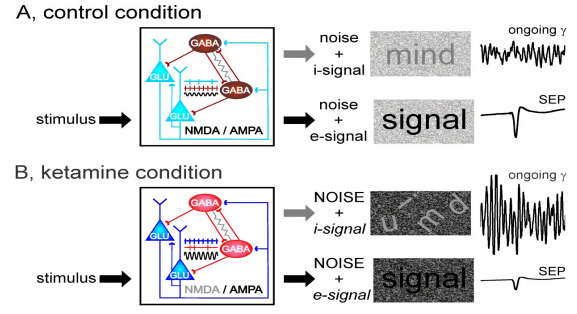
Figures(2)

Didier Pinault. N-Methyl D-Aspartate Receptor Antagonists Amplify Network Baseline Gamma Frequency (30–80 Hz) Oscillations: Noise and Signal[J]. AIMS Neuroscience, 2014, 1(2): 169-182. doi: 10.3934/Neuroscience.2014.2.169







 DownLoad:
DownLoad: