Hereditary Hemochromatosis (HH) is an autosomal recessive disorder of iron metabolism associated with HFE gene mutations, characterized by increased iron absorption and accumulation leading to multi-organ damage caused by iron overload toxicity. Beta thalassemia is caused by a mutation in the human beta globin gene. Imbalanced production of globin chain results in beta thalassemia, where the unpaired alpha chains precipitates in red cell precursors leading to ineffective erythropoiesis and reduced RBC survival. Both HH and beta thalassemia condition results in rapid accumulation of iron lead to iron overload in tissues and organs. The study aims to analyze the frequency of HFE variants among beta thalassemia cases and their effect on iron overload. The frequency of three HFE variants C282Y, H63D, S65C was analyzed by PCR RFLP method among Beta Thalassemia Trait (BTT) (n = 203), Beta Thalassemia Major (BTM) (n = 19) and age and sex-matched control samples (n = 200). The present study furnished allele frequency of H63D variant in BTT, BTM and controls 8.13, 15.8 and 6% respectively. Ten out of 33 heterozygous H63D variants exhibited iron overload with higher ferritin levels indicating HFE variant might aggravate the absorption of iron. The C282Y variant was present in heterozygous state in 1 case among beta thalassemia carriers. The C282Y variant was absent among BTM and control cases. S65C HFE variant was absent in the present study. Iron overload was completely absent in the control cases among all three HFE genotypes. Hence it is inferred from the present investigation, analysis of HFE genes and iron status will remarkably help to reason out the probable reason behind the iron status and support in proper management of beta thalassemia cases.
Citation: Bhuvana Selvaraj, Sangeetha Soundararajan, Shettu Narayanasamy, Ganesan Subramanian, Senthil Kumar Ramanathan. Frequency of hereditary hemochromatosis gene mutations and their effects on iron overload among beta thalassemia patients of Chennai residents[J]. AIMS Molecular Science, 2021, 8(4): 233-247. doi: 10.3934/molsci.2021018
Hereditary Hemochromatosis (HH) is an autosomal recessive disorder of iron metabolism associated with HFE gene mutations, characterized by increased iron absorption and accumulation leading to multi-organ damage caused by iron overload toxicity. Beta thalassemia is caused by a mutation in the human beta globin gene. Imbalanced production of globin chain results in beta thalassemia, where the unpaired alpha chains precipitates in red cell precursors leading to ineffective erythropoiesis and reduced RBC survival. Both HH and beta thalassemia condition results in rapid accumulation of iron lead to iron overload in tissues and organs. The study aims to analyze the frequency of HFE variants among beta thalassemia cases and their effect on iron overload. The frequency of three HFE variants C282Y, H63D, S65C was analyzed by PCR RFLP method among Beta Thalassemia Trait (BTT) (n = 203), Beta Thalassemia Major (BTM) (n = 19) and age and sex-matched control samples (n = 200). The present study furnished allele frequency of H63D variant in BTT, BTM and controls 8.13, 15.8 and 6% respectively. Ten out of 33 heterozygous H63D variants exhibited iron overload with higher ferritin levels indicating HFE variant might aggravate the absorption of iron. The C282Y variant was present in heterozygous state in 1 case among beta thalassemia carriers. The C282Y variant was absent among BTM and control cases. S65C HFE variant was absent in the present study. Iron overload was completely absent in the control cases among all three HFE genotypes. Hence it is inferred from the present investigation, analysis of HFE genes and iron status will remarkably help to reason out the probable reason behind the iron status and support in proper management of beta thalassemia cases.
Beta Thalassemia Homozygous
Beta Thalassemia Major
Beta Thalassemia Trait
Hemoglobin Subunit Beta gene
Hereditary Hemochromatosis
Single Nucleotide Polymorphism
Wild type allele

| [1] |
Martins R, Picanço I, Fonseca A, et al. (2004) The role of HFE mutations on iron metabolism in beta-thalassemia carriers. J Hum Genet 49: 651-655. doi: 10.1007/s10038-004-0202-z

|
| [2] | Hash RB (2001) Hereditary hemochromatosis. J Am Board Fam Pract 14: 266-273. |
| [3] |
Mura C, Raguenes O, Férec C (1999) HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 93: 2502-2505. doi: 10.1182/blood.V93.8.2502
|
| [4] | Pietrangelo A, Camaschella C (1998) Molecular genetics and control of iron metabolism in hemochromatosis. Haematologica 83: 456-461. |
| [5] |
Agarwal S, Tewari D, Arya V, et al. (2007) Status of HFE mutation in thalassemia syndromes in north India. Ann Hematol 86: 483-485. doi: 10.1007/s00277-006-0224-z
|
| [6] |
Hanson EH, Imperatore G, Burke W (2001) HFE gene and hereditary hemochromatosis: a HuGE review. Am J Epidemiol 154: 193-206. doi: 10.1093/aje/154.3.193
|
| [7] |
Weatherall DJ, Clegg JB (2001) The Thalassemia Syndromes Oxford: Blackwell Science Ltd. doi: 10.1002/9780470696705
|
| [8] |
Chaudhary S, Dhawan D, Sojitra N, et al. (2017) Whole Gene Sequencing Based Screening Approach to Detect β-Thalassemia Mutations. Biol Med 9: 383. doi: 10.4172/0974-8369.1000383
|
| [9] | Mokhtar DA, Hamdy MM, Badr AM (2013) Frequency of human hemochromatosis (HFE) gene mutations in Egyptians with β-thalassemia. Egypt J Haematol 38: 36. |
| [10] | Kaur G, Rapthap CC, Xavier M, et al. (2003) Distribution of C282Y and H63D mutations in the HFE gene in healthy Asian Indians and patients with thalassaemia major. Natl Med J India 16: 309-310. |
| [11] | Čimburová M, Půtová I, Provaznikova H, et al. (2005) S65C and other mutations in the haemochromatosis gene in the Czech population. Folia Biologica (Praha) 51: 172-176. |
| [12] |
Gomez-Llorente C, Miranda-León MT, Blanco S, et al. (2005) Frequency and clinical expression of HFE gene mutations in a Spanish population of subjects with abnormal iron metabolism. Ann Hematol 84: 650-655. doi: 10.1007/s00277-005-1069-6
|
| [13] |
Cukjati M, Vaupotič T, Rupreht R, et al. (2007) Prevalence of H63D, S65C and C282Y hereditary hemochromatosis gene mutations in Slovenian population by an improved high-throughput genotyping assay. BMC Med Genet 8: 69. doi: 10.1186/1471-2350-8-69
|
| [14] |
Njajou OT, Houwing-Duistermaat JJ, Osborne RH, et al. (2003) A population-based study of the effect of the HFE C282Y and H63D mutations on iron metabolism. Eur J Hum Genet 11: 225. doi: 10.1038/sj.ejhg.5200955
|
| [15] | Piperno A, Mariani R, Arosio C, et al. (2000) Haemochromatosis in patients with β-thalassaemia trait. Br J Haematol 111: 908-914. |
| [16] |
Trent J, Le H, Yu B, et al. (2000) DNA testing for haemochromatosis: diagnostic, predictive and screening implications. Pathology 32: 274-279. doi: 10.1080/pat.32.4.274.279
|
| [17] |
Madani HA, Afify RA, Abd El-Aal AA, et al. (2011) Role of HFE gene mutations on developing iron overload in β-thalassaemia carriers in Egypt. East Mediterr Health J 17: 546-551. doi: 10.26719/2011.17.6.546
|
| [18] |
Enein AA, El Dessouky NA, Mohamed KS, et al. (2016) Frequency of hereditary hemochromatosis (HFE) gene mutations in Egyptian beta thalassemia patients and its relation to iron overload. Open Access Maced J Med Sci 4: 226. doi: 10.3889/oamjms.2016.055
|
| [19] |
Wilson MM, Al-Wakeel H, Said F, et al. (2015) Study of the effect of HFE gene mutations on iron overload in Egyptian thalassemia patients. Egypt J Med Hum Genet 16: 129-133. doi: 10.1016/j.ejmhg.2015.02.002
|
| [20] | Elmrghni S, Dixon RA, Williams DR (2011) Frequencies of HFE gene mutations associated with haemochromatosis in the population of Libya living in Benghazi. Int J Clin Exp Med 4: 200. |
| [21] | Ali N, Ahmed B, Akram H, et al. (2018) HFE gene mutations. Prof Med J 25: 129-134. |
| [22] | Soltanpour MS, Farshdousti Hagh M, Kamali K, et al. (2017) Frequency of C282Y and H63D Mutations of HFE Gene and Their Correlation with Iron Status in Iranian Beta-Thalassemia Major Patients. Iran J Pediatr Hematol Oncol 7: 154-162. |
| [23] |
Tiwari AK, Behera TR, Kujur M, et al. (2016) Association of HFE gene mutation in thalassemia major patients. J Evid Based Med Healthc 3: 4853-5485. doi: 10.18410/jebmh/2016/1022
|
| [24] |
Dhillon BK, Das R, Garewal G, et al. (2007) Frequency of primary iron overload and HFE gene mutations (C282Y, H63D and S65C) in chronic liver disease patients in north India. World J Gastroenterol 13: 2956. doi: 10.3748/wjg.v13.i21.2956
|
| [25] |
Nadkarni AH, Singh AA, Colaco S, et al. (2017) Effect of the Hemochromatosis Mutations on Iron Overload among the Indian β Thalassemia Carriers. J Clin Lab Anal 31: e22054. doi: 10.1002/jcla.22054
|
| [26] |
Dasgupta A, Roy S, Sinharay M (2017) Prevalance of H63D and C282Y Mutation and Its Association with Iron Overload in Thalassemia Patients in an Eastern Indian Population. J Adv Med Med Res 1-10. doi: 10.9734/JAMMR/2017/35374
|
| [27] | El-Rashidi FH, Elshafey AE, Ragab SM, et al. (2008) Haemochromatosis gene mutation H63D is a risk factor for iron overload in Egyptian beta-thalassemic children. Egypt J Med Hum Genet 9: 149-160. |
| [28] |
Steinberg KK, Cogswell ME, Chang JC, et al. (2001) Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. Jama 285: 2216-2222. doi: 10.1001/jama.285.17.2216
|
| [29] | Herkenhoff ME, Pitlovanciv AK, Remualdo VR (2016) Prevalence of C282Y and H63D mutations in the HFEgene in patients from São Paulo and Southern Brazil. J Bras Patol Med Lab 52: 21-24. |
| [30] |
Oliveira TM, Souza FP, Jardim ACG, et al. (2006) HFE gene mutations in Brazilian thalassemic patients. Braz J Med Biol Res 39: 1575-1580. doi: 10.1590/S0100-879X2006001200008
|
| [31] | Leão GDR, Oliveira TMM, Fernandes TMM, et al. (2010) Analysis of C282Y and H63D mutations of the HFE gene in patients with persistent hyperferritinemia. J Med Med Sci 1: 453-459. |
| [32] | Bain BJ (2006) Haemoglobinopathy Diagnosis Oxford, UK: Blackwell, 1-314. |
| [33] | Fekri K, Rasouli NA, Zavareh SAT, et al. (2019) Hepcidin and HFE polymorphisms and ferritin level in β-Thalassemia major. Int J Hematol Oncol Stem Cell Res 13: 42-48. |
| [34] | Melis MA, Cau M, Deidda F, et al. (2002) H63D mutation in the HFE gene increases iron overload in beta-thalassemia carriers. Haematologica 87: 242-245. |

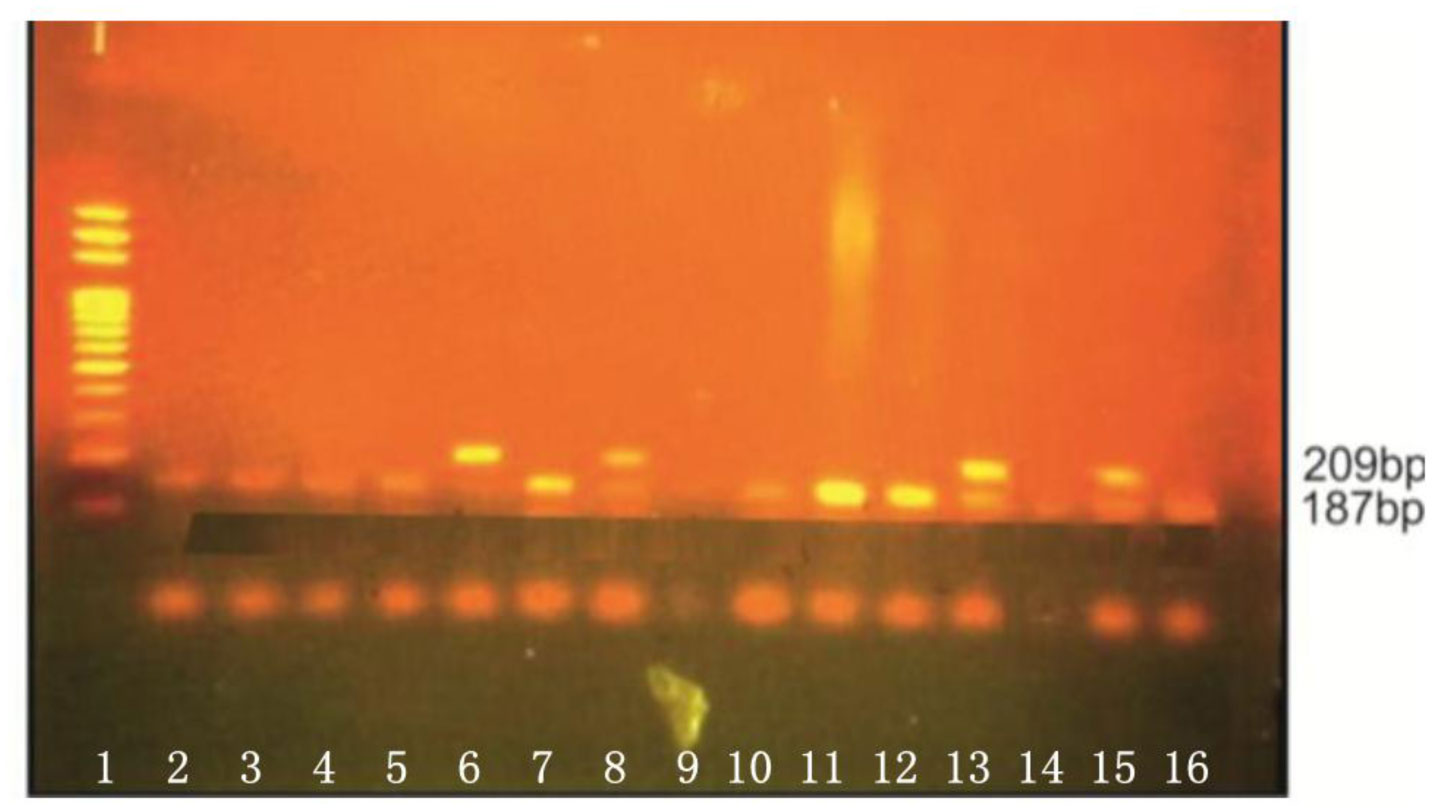
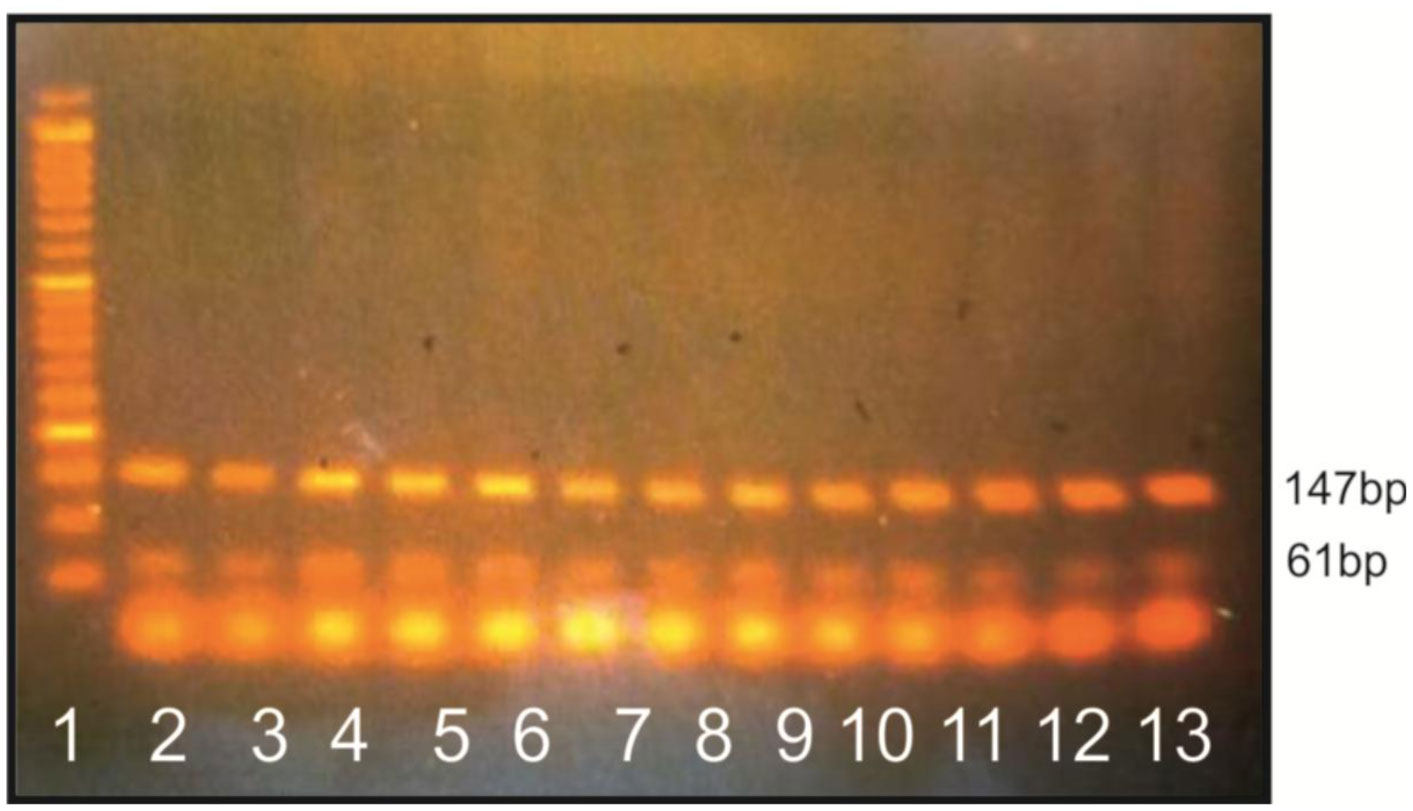
Figures(3) / Tables(5)

Bhuvana Selvaraj, Sangeetha Soundararajan, Shettu Narayanasamy, Ganesan Subramanian, Senthil Kumar Ramanathan. Frequency of hereditary hemochromatosis gene mutations and their effects on iron overload among beta thalassemia patients of Chennai residents[J]. AIMS Molecular Science, 2021, 8(4): 233-247. doi: 10.3934/molsci.2021018







 DownLoad:
DownLoad: