Citation: Neha Sinha, Mark A. Seeley, Daniel S. Horwitz, Hemil Maniar, Andrea H. Seeley. Pediatric Orthogenomics: The Latest Trends and Controversies[J]. AIMS Medical Science, 2017, 4(2): 192-216. doi: 10.3934/medsci.2017.2.192

| [1] |
Eknoyan G (2006) On the origin of genetics and beginnings of medical genetics of diseases of the kidney. Adv Chronic Kidney Dis 13: 174-177. doi: 10.1053/j.ackd.2006.01.004

|
| [2] | Keller EF (2002, Print) The Century of the Gene. Cambridge, MA: Harvard UP, 2002. |
| [3] |
Portin P (2014) The birth and development of the DNA theory of inheritance: sixty years since the discovery of the structure of DNA. J Genet 93: 293-302. doi: 10.1007/s12041-014-0337-4
|
| [4] |
Mullis KB (1990) The unusual origin of the polymerase chain reaction. Sci Am 262: 56-61, 64-5. doi: 10.1038/scientificamerican0490-56
|
| [5] | Sweeney BP (2004) Watson and Crick 50 years on. From double helix to pharmacogenomics. Anaesthesia 59: 150-165. |
| [6] | Evans CH, Rosier RN (2005) Molecular biology in orthopaedics: the advent of molecular orthopaedics. J Bone Joint Surg Am 87: 2550-2564. |
| [7] | Puzas JE, O'Keefe RJ, Lieberman JR (2002) The orthopaedic genome: what does the future hold and are we ready?. J Bone Joint Surg Am 84-A: 133-141. |
| [8] | Bayat A, Barton A, Ollier WE (2004) Dissection of complex genetic disease: implications for orthopaedics. Clin Orthop Relat Res (419): 297-305. |
| [9] |
Matzko ME, Bowen TR, Smith WR (2012) Orthogenomics: an update. J Am Acad Orthop Surg 20: 536-546. doi: 10.5435/JAAOS-20-08-536
|
| [10] |
Riegel M (2014) Human molecular cytogenetics: From cells to nucleotides. Genet Mol Biol 37: 194-209. doi: 10.1590/S1415-47572014000200006
|
| [11] |
Langer-Safer PR, Levine M, Ward DC (1982) Immunological method for mapping genes on Drosophila polytene chromosomes. Proc Natl Acad Sci U S A 79: 4381-4385. doi: 10.1073/pnas.79.14.4381
|
| [12] |
Kallioniemi A, Kallioniemi OP, Sudar D, et al. (1992) Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258: 818-821. doi: 10.1126/science.1359641
|
| [13] |
Pinkel D, Segraves R, Sudar D, et al. (1998) High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20: 207-211. doi: 10.1038/2524
|
| [14] |
Solinas-Toldo S, Lampel S, Stilgenbauer S, et al. (1997) Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20: 399-407. doi: 10.1002/(SICI)1098-2264(199712)20:4<399::AID-GCC12>3.0.CO;2-I
|
| [15] |
Wiszniewska J, Bi W, Shaw C, et al. (2014) Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet 22: 79-87. doi: 10.1038/ejhg.2013.77
|
| [16] |
Shashi V, McConkie-Rosell A, Rosell B, et al. (2014) The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 16: 176-182. doi: 10.1038/gim.2013.99
|
| [17] |
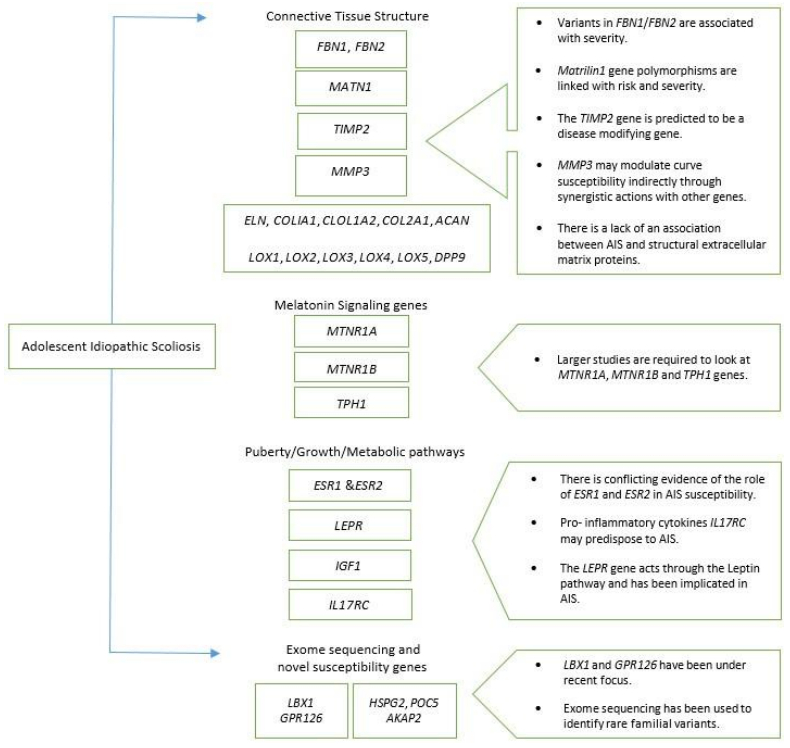
Ogilvie J (2010) Adolescent idiopathic scoliosis and genetic testing. Curr Opin Pediatr 22: 67-70. doi: 10.1097/MOP.0b013e32833419ac
|
| [18] | Horne JP, Flannery R, Usman S (2014) Adolescent idiopathic scoliosis: diagnosis and management. Am Fam Physician 89: 193-198. |
| [19] |
Riseborough EJ, Wynne-Davies R (1973) A genetic survey of idiopathic scoliosis in Boston, Massachusetts. J Bone Joint Surg Am 55: 974-982. doi: 10.2106/00004623-197355050-00006
|
| [20] | Kesling KL, Reinker KA (1997) Scoliosis in twins. A meta-analysis of the literature and report of six cases. Spine (Phila Pa 1976) 22: 2009-2014; |
| [21] |
Wu J, Qiu Y, Zhang L, et al. (2006) Association of estrogen receptor gene polymorphisms with susceptibility to adolescent idiopathic scoliosis. Spine (Phila Pa 1976) 31: 1131-1136. doi: 10.1097/01.brs.0000216603.91330.6f
|
| [22] |
Chen S, Zhao L, Roffey DM, et al. (2014) Association between the ESR1-351A > G single nucleotide polymorphism (rs9340799) and adolescent idiopathic scoliosis: a systematic review and meta-analysis. Eur Spine J 23: 2586-2593. doi: 10.1007/s00586-014-3481-x
|
| [23] | Zhao L, Roffey DM, Chen S (2016) Association between the Estrogen Receptor Beta (ESR2) Rs1256120 Single Nucleotide Polymorphism and Adolescent Idiopathic Scoliosis: A Systematic Review and Meta-Analysis. Spine (Phila Pa 1976): Epub ahead of print. |
| [24] | Yang P, Liu H, Lin J, et al. (2015) The Association of rs4753426 Polymorphism in the Melatonin Receptor 1B (MTNR1B) Gene and Susceptibility to Adolescent Idiopathic Scoliosis: A Systematic Review and Meta-analysis. Pain Physician 18: 419-431. |
| [25] |
Ogura Y, Kou I, Miura S, et al. (2015) A Functional SNP in BNC2 Is Associated with Adolescent Idiopathic Scoliosis. Am J Hum Genet 97: 337-342. doi: 10.1016/j.ajhg.2015.06.012
|
| [26] |
Buchan JG, Alvarado DM, Haller GE, et al. (2014) Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum Mol Genet 23: 5271-5282. doi: 10.1093/hmg/ddu224
|
| [27] |
Liu Z, Wang F, Xu LL, et al. (2015) Polymorphism of rs2767485 in Leptin Receptor Gene is Associated With the Occurrence of Adolescent Idiopathic Scoliosis. Spine (Phila Pa 1976) 40: 1593-1598. doi: 10.1097/BRS.0000000000001095
|
| [28] |
Zhou S, Qiu XS, Zhu ZZ, et al. (2012) A single-nucleotide polymorphism rs708567 in the IL-17RC gene is associated with a susceptibility to and the curve severity of adolescent idiopathic scoliosis in a Chinese Han population: a case-control study. BMC Musculoskelet Disord 13: 181-2474-13-181. doi: 10.1186/1471-2474-13-181
|
| [29] |
Ryzhkov II, Borzilov EE, Churnosov MI, et al. (2013) Transforming growth factor beta 1 is a novel susceptibility gene for adolescent idiopathic scoliosis. Spine (Phila Pa 1976) 38: E699-704. doi: 10.1097/BRS.0b013e31828de9e1
|
| [30] |
Zhang H, Zhao S, Zhao Z, et al. (2014) The association of rs1149048 polymorphism in matrilin-1(MATN1) gene with adolescent idiopathic scoliosis susceptibility: a meta-analysis. Mol Biol Rep 41: 2543-2549. doi: 10.1007/s11033-014-3112-y
|
| [31] |
Bae JW, Cho CH, Min WK, et al. (2012) Associations between matrilin-1 gene polymorphisms and adolescent idiopathic scoliosis curve patterns in a Korean population. Mol Biol Rep 39: 5561-5567. doi: 10.1007/s11033-011-1360-7
|
| [32] | Yu Y, Chen ZJ, Qiu Y, et al. (2009) Association between matrilin-1 gene polymorphism and bracing effectiveness in adolescent idiopathic scoliosis girls. Zhonghua Wai Ke Za Zhi 47: 1728-1731. |
| [33] | Wang B, Chen ZJ, Qiu Y, et al. (2009) Decreased circulating matrilin-1 levels in adolescent idiopathic scoliosis. Zhonghua Wai Ke Za Zhi 47: 1638-1641. |
| [34] | Chen ZJ, Qiu Y, Yu Y, et al. (2009) Association between polymorphism of Matrilin-1 gene (MATN1) with susceptibility to adolescent idiopathic scoliosis. Zhonghua Wai Ke Za Zhi 47: 1332-1335. |
| [35] |
Montanaro L, Parisini P, Greggi T, et al. (2006) Evidence of a linkage between matrilin-1 gene (MATN1) and idiopathic scoliosis. Scoliosis 1: 21. doi: 10.1186/1748-7161-1-21
|
| [36] |
Wang H, Wu Z, Zhuang Q, et al. (2008) Association study of tryptophan hydroxylase 1 and arylalkylamine N-acetyltransferase polymorphisms with adolescent idiopathic scoliosis in Han Chinese. Spine (Phila Pa 1976) 33: 2199-2203. doi: 10.1097/BRS.0b013e31817c03f9
|
| [37] |
Gorman KF, Julien C, Moreau A (2012) The genetic epidemiology of idiopathic scoliosis. Eur Spine J 21: 1905-1919. doi: 10.1007/s00586-012-2389-6
|
| [38] | Zhu Z, Xu L, Qiu Y (2015) Current progress in genetic research of adolescent idiopathic scoliosis. Ann Transl Med 3: S19. |
| [39] |
Pearson TA, Manolio TA (2008) How to interpret a genome-wide association study. JAMA 299: 1335-1344. doi: 10.1001/jama.299.11.1335
|
| [40] |
Chettier R, Nelson L, Ogilvie JW, et al. (2015) Haplotypes at LBX1 have distinct inheritance patterns with opposite effects in adolescent idiopathic scoliosis. PLoS One 10: e0117708. doi: 10.1371/journal.pone.0117708
|
| [41] |
Ikegawa S (2016) Genomic study of adolescent idiopathic scoliosis in Japan. Scoliosis Spinal Disord 11: 5-016-0067-x. doi: 10.1186/s13013-016-0067-x
|
| [42] |
Grauers A, Wang J, Einarsdottir E, et al. (2015) Candidate gene analysis and exome sequencing confirm LBX1 as a susceptibility gene for idiopathic scoliosis. Spine J 15: 2239-2246. doi: 10.1016/j.spinee.2015.05.013
|
| [43] |
Jagla K, Dolle P, Mattei MG, et al. (1995) Mouse Lbx1 and human LBX1 define a novel mammalian homeobox gene family related to the Drosophila lady bird genes. Mech Dev 53: 345-356. doi: 10.1016/0925-4773(95)00450-5
|
| [44] | Gross MK, Moran-Rivard L, Velasquez T, et al. (2000) Lbx1 is required for muscle precursor migration along a lateral pathway into the limb. Development 127: 413-424. |
| [45] |
Schafer K, Neuhaus P, Kruse J, et al. (2003) The homeobox gene Lbx1 specifies a subpopulation of cardiac neural crest necessary for normal heart development. Circ Res 92: 73-80. doi: 10.1161/01.RES.0000050587.76563.A5
|
| [46] |
Gross MK, Dottori M, Goulding M (2002) Lbx1 specifies somatosensory association interneurons in the dorsal spinal cord. Neuron 34: 535-549. doi: 10.1016/S0896-6273(02)00690-6
|
| [47] |
Xu JF, Yang GH, Pan XH, et al. (2015) Association of GPR126 gene polymorphism with adolescent idiopathic scoliosis in Chinese populations. Genomics 105: 101-107. doi: 10.1016/j.ygeno.2014.11.009
|
| [48] |
Kou I, Takahashi Y, Johnson TA, et al. (2013) Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat Genet 45: 676-679. doi: 10.1038/ng.2639
|
| [49] | Zhao L, Roffey DM, Chen S (2015) Genetics of adolescent idiopathic scoliosis in the post-genome-wide association study era. Ann Transl Med 3: S35. |
| [50] |
Stankiewicz P, Lupski JR (2010) Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437-455. doi: 10.1146/annurev-med-100708-204735
|
| [51] |
Buchan JG, Alvarado DM, Haller G, et al. (2014) Are copy number variants associated with adolescent idiopathic scoliosis?. Clin Orthop Relat Res 472: 3216-3225. doi: 10.1007/s11999-014-3766-8
|
| [52] |
Costell M, Gustafsson E, Aszodi A, et al. (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 147: 1109-1122. doi: 10.1083/jcb.147.5.1109
|
| [53] |
Rodgers KD, Sasaki T, Aszodi A, et al. (2007) Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum Mol Genet 16: 515-528. doi: 10.1093/hmg/ddl484
|
| [54] |
Stum M, Davoine CS, Vicart S, et al. (2006) Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum Mutat 27: 1082-1091. doi: 10.1002/humu.20388
|
| [55] | Baschal EE, Wethey CI, Swindle K, et al. (2014) Exome sequencing identifies a rare HSPG2 variant associated with familial idiopathic scoliosis. G3 (Bethesda) 5: 167-174. |
| [56] |
Robinson PN, Godfrey M (2000) The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet 37: 9-25. doi: 10.1136/jmg.37.1.9
|
| [57] |
Tuncbilek E, Alanay Y (2006) Congenital contractural arachnodactyly (Beals syndrome). Orphanet J Rare Dis 1: 20. doi: 10.1186/1750-1172-1-20
|
| [58] | Patten SA, Margaritte-Jeannin P, Bernard JC, et al. (2015) Functional variants of POC5 identified in patients with idiopathic scoliosis. J Clin Invest 125: 1124-1128. |
| [59] |
Li W, Li Y, Zhang L, et al. (2016) AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J Med Genet 53: 488-493. doi: 10.1136/jmedgenet-2015-103684
|
| [60] |
Weinstein SL, Dolan LA, Wright JG, et al. (2013) Effects of bracing in adolescents with idiopathic scoliosis. N Engl J Med 369: 1512-1521. doi: 10.1056/NEJMoa1307337
|
| [61] |
Ward K, Ogilvie JW, Singleton MV, et al. (2010) Validation of DNA-based prognostic testing to predict spinal curve progression in adolescent idiopathic scoliosis. Spine (Phila Pa 1976) 35: E1455-1464. doi: 10.1097/BRS.0b013e3181ed2de1
|
| [62] |
Roye BD, Wright ML, Matsumoto H, et al. (2015) An Independent Evaluation of the Validity of a DNA-Based Prognostic Test for Adolescent Idiopathic Scoliosis. J Bone Joint Surg Am 97: 1994-1998. doi: 10.2106/JBJS.O.00217
|
| [63] | Lee MC (2015) The Distance from Bench to Bedside: Commentary on an article by Benjamin D. Roye, MD, MPH, et al..: "An Independent Evaluation of the Validity of a DNA-Based Prognostic Test for Adolescent Idiopathic Scoliosis". J Bone Joint Surg Am 97: e79. |
| [64] |
Tang QL, Julien C, Eveleigh R, et al. (2015) A replication study for association of 53 single nucleotide polymorphisms in ScoliScore test with adolescent idiopathic scoliosis in French-Canadian population. Spine (Phila Pa 1976) 40: 537-543. doi: 10.1097/BRS.0000000000000807
|
| [65] | Bohl DD, Telles CJ, Ruiz FK, et al. (2016) A Genetic Test Predicts Providence Brace Success for Adolescent Idiopathic Scoliosis When Failure Is Defined as Progression to >45 Degrees. Clin Spine Surg 29: E146-50. |
| [66] |
Xu L, Qiu X, Sun X, et al. (2011) Potential genetic markers predicting the outcome of brace treatment in patients with adolescent idiopathic scoliosis. Eur Spine J 20: 1757-1764. doi: 10.1007/s00586-011-1874-7
|
| [67] |
Lowry RB, Bedard T (2016) Congenital limb deficiency classification and nomenclature: The need for a consensus. Am J Med Genet A 170: 1400-1404. doi: 10.1002/ajmg.a.37608
|
| [68] | Gold NB, Westgate MN, Holmes LB (2011) Anatomic and etiological classification of congenital limb deficiencies. Am J Med Genet A 155A: 1225-1235. |
| [69] | Auerbach AD, Allen RG (1991) Leukemia and preleukemia in Fanconi anemia patients. A review of the literature and report of the International Fanconi Anemia Registry. Cancer Genet Cytogenet 51: 1-12. |
| [70] |
Hurst JA, Hall CM, Baraitser M (1991) The Holt-Oram syndrome. J Med Genet 28: 406-410. doi: 10.1136/jmg.28.6.406
|
| [71] |
Hall JG (1987) Thrombocytopenia and absent radius (TAR) syndrome. J Med Genet 24: 79-83. doi: 10.1136/jmg.24.2.79
|
| [72] | Barham G, Clarke NM (2008) Genetic regulation of embryological limb development with relation to congenital limb deformity in humans. J Child Orthop 2: 1-9. |
| [73] |
Zuniga A, Zeller R, Probst S (2012) The molecular basis of human congenital limb malformations. Wiley Interdiscip Rev Dev Biol 1: 803-822. doi: 10.1002/wdev.59
|
| [74] | Wang YH, Keenan SR, Lynn J, et al. (2015) Gremlin1 induces anterior-posterior limb bifurcations in developing Xenopus limbs but does not enhance limb regeneration. Mech Dev 138 Pt 3: 256-267. |
| [75] | Amprino R, Bonetti DA (1967) Experimental observations in the development of ectoderm-free mesoderm of the limb bud in chick embryos. Nature 214: 826-827. |
| [76] |
Brewer JR, Mazot P, Soriano P (2016) Genetic insights into the mechanisms of Fgf signaling. Genes Dev 30: 751-771. doi: 10.1101/gad.277137.115
|
| [77] |
Manouvrier-Hanu S, Holder-Espinasse M, Lyonnet S (1999) Genetics of limb anomalies in humans. Trends Genet 15: 409-417. doi: 10.1016/S0168-9525(99)01823-5
|
| [78] |
Sun X, Mariani FV, Martin GR (2002) Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature 418: 501-508. doi: 10.1038/nature00902
|
| [79] |
Boulet AM, Moon AM, Arenkiel BR, et al. (2004) The roles of Fgf4 and Fgf8 in limb bud initiation and outgrowth. Dev Biol 273: 361-372. doi: 10.1016/j.ydbio.2004.06.012
|
| [80] |
Zeller R, Zuniga A (2007) Shh and Gremlin1 chromosomal landscapes in development and disease. Curr Opin Genet Dev 17: 428-434. doi: 10.1016/j.gde.2007.07.006
|
| [81] |
Khokha MK, Hsu D, Brunet LJ, et al. (2003) Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat Genet 34: 303-307. doi: 10.1038/ng1178
|
| [82] |
Dimitrov BI, Voet T, De Smet L, et al. (2010) Genomic rearrangements of the GREM1-FMN1 locus cause oligosyndactyly, radio-ulnar synostosis, hearing loss, renal defects syndrome and Cenani--Lenz-like non-syndromic oligosyndactyly. J Med Genet 47: 569-574. doi: 10.1136/jmg.2009.073833
|
| [83] |
Gong Y, Krakow D, Marcelino J, et al. (1999) Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 21: 302-304. doi: 10.1038/6821
|
| [84] |
Walsh DW, Godson C, Brazil DP, et al. (2010) Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol 20: 244-256. doi: 10.1016/j.tcb.2010.01.008
|
| [85] | Garavelli L, Wischmeijer A, Rosato S, et al. (2011) Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome and WNT7A mutations: genetic homogeneity and nosological delineation. Am J Med Genet A 155A: 332-336. |
| [86] |
Mortlock DP, Innis JW (1997) Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet 15: 179-180. doi: 10.1038/ng0297-179
|
| [87] |
Goodman FR (2002) Limb malformations and the human HOX genes. Am J Med Genet 112: 256-265. doi: 10.1002/ajmg.10776
|
| [88] |
Duboc V, Logan MP (2011) Regulation of limb bud initiation and limb-type morphology. Dev Dyn 240: 1017-1027. doi: 10.1002/dvdy.22582
|
| [89] | King M, Arnold JS, Shanske A, et al. (2006) T-genes and limb bud development. Am J Med Genet A 140: 1407-1413. |
| [90] |
Liu C, Nakamura E, Knezevic V, et al. (2003) A role for the mesenchymal T-box gene Brachyury in AER formation during limb development. Development 130: 1327-1337. doi: 10.1242/dev.00354
|
| [91] |
Bamshad M, Lin RC, Law DJ, et al. (1997) Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet 16: 311-315. doi: 10.1038/ng0797-311
|
| [92] |
Davenport TG, Jerome-Majewska LA, Papaioannou VE (2003) Mammary gland, limb and yolk sac defects in mice lacking Tbx3, the gene mutated in human ulnar mammary syndrome. Development 130: 2263-2273. doi: 10.1242/dev.00431
|
| [93] |
Rallis C, Del Buono J, Logan MP (2005) Tbx3 can alter limb position along the rostrocaudal axis of the developing embryo. Development 132: 1961-1970. doi: 10.1242/dev.01787
|
| [94] |
Don EK, de Jong-Curtain TA, Doggett K, et al. (2016) Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol Open 5: 359-366. doi: 10.1242/bio.016295
|
| [95] |
Ahn DG, Kourakis MJ, Rohde LA, et al. (2002) T-box gene tbx5 is essential for formation of the pectoral limb bud. Nature 417: 754-758. doi: 10.1038/nature00814
|
| [96] |
Kiefer SM, Robbins L, Barina A, et al. (2008) SALL1 truncated protein expression in Townes-Brocks syndrome leads to ectopic expression of downstream genes. Hum Mutat 29: 1133-1140. doi: 10.1002/humu.20759
|
| [97] |
Kohlhase J, Wischermann A, Reichenbach H, et al. (1998) Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet 18: 81-83. doi: 10.1038/ng0198-81
|
| [98] | Al-Qattan MM (2011) WNT pathways and upper limb anomalies. J Hand Surg Eur Vol 36: 9-22. |
| [99] |
Sowinska-Seidler A, Socha M, Jamsheer A (2014) Split-hand/foot malformation-molecular cause and implications in genetic counseling. J Appl Genet 55: 105-115. doi: 10.1007/s13353-013-0178-5
|
| [100] |
Naveed M, Nath SK, Gaines M, et al. (2007) Genomewide linkage scan for split-hand/foot malformation with long-bone deficiency in a large Arab family identifies two novel susceptibility loci on chromosomes 1q42.2-q43 and 6q14.1. Am J Hum Genet 80: 105-111. doi: 10.1086/510724
|
| [101] | Gurnett CA, Dobbs MB, Nordsieck EJ, et al. (2006) Evidence for an additional locus for split hand/foot malformation in chromosome region 8q21.11-q22.3. Am J Med Genet A 140: 1744-1748. |
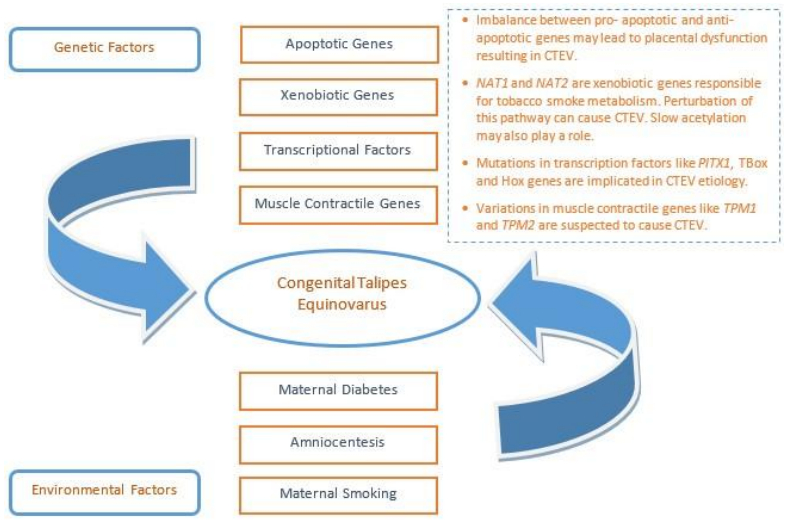
| [102] | Jiang B, Zhang Z, Zheng P, et al. (2014) Apoptotic genes expression in placenta of clubfoot-like fetus pregnant rats. Int J Clin Exp Pathol 7: 677-684. |
| [103] |
Alderman BW, Takahashi ER, LeMier MK (1991) Risk indicators for talipes equinovarus in Washington State, 1987-1989. Epidemiology 2: 289-292. doi: 10.1097/00001648-199107000-00009
|
| [104] | Chung CS, Nemechek RW, Larsen IJ, et al. (1969) Genetic and epidemiological studies of clubfoot in Hawaii. General and medical considerations. Hum Hered 19: 321-342. |
| [105] |
Moorthi RN, Hashmi SS, Langois P, et al. (2005) Idiopathic talipes equinovarus (ITEV) (clubfeet) in Texas. Am J Med Genet A 132A: 376-380. doi: 10.1002/ajmg.a.30505
|
| [106] |
Miedzybrodzka Z (2003) Congenital talipes equinovarus (clubfoot): a disorder of the foot but not the hand. J Anat 202: 37-42. doi: 10.1046/j.1469-7580.2003.00147.x
|
| [107] |
Irani RN, Sherman MS (1972) The pathological anatomy of idiopathic clubfoot. Clin Orthop Relat Res 84: 14-20. doi: 10.1097/00003086-197205000-00004
|
| [108] |
Bacino CA, Hecht JT (2014) Etiopathogenesis of equinovarus foot malformations. Eur J Med Genet 57: 473-479. doi: 10.1016/j.ejmg.2014.06.001
|
| [109] |
Parker SE, Mai CT, Strickland MJ, et al. (2009) Multistate study of the epidemiology of clubfoot. Birth Defects Res A Clin Mol Teratol 85: 897-904. doi: 10.1002/bdra.20625
|
| [110] |
Rogers JM (2009) Tobacco and pregnancy. Reprod Toxicol 28: 152-160. doi: 10.1016/j.reprotox.2009.03.012
|
| [111] |
Lambers DS, Clark KE (1996) The maternal and fetal physiologic effects of nicotine. Semin Perinatol 20: 115-126. doi: 10.1016/S0146-0005(96)80079-6
|
| [112] |
Hecht JT, Ester A, Scott A, et al. (2007) NAT2 variation and idiopathic talipes equinovarus (clubfoot). Am J Med Genet A 143A: 2285-2291. doi: 10.1002/ajmg.a.31927
|
| [113] |
Sommer A, Blanton SH, Weymouth K, et al. (2011) Smoking, the xenobiotic pathway, and clubfoot. Birth Defects Res A Clin Mol Teratol 91: 20-28. doi: 10.1002/bdra.20742
|
| [114] | 114. Engell V, Damborg F, Andersen M, et al. (2006) Club foot: a twin study. J Bone Joint Surg Br 88: 374-376. |
| [115] |
de Andrade M, Barnholtz JS, Amos CI, et al. (1998) Segregation analysis of idiopathic talipes equinovarus in a Texan population. Am J Med Genet 79: 97-102. doi: 10.1002/(SICI)1096-8628(19980901)79:2<97::AID-AJMG4>3.0.CO;2-K
|
| [116] |
Honein MA, Paulozzi LJ, Moore CA (2000) Family history, maternal smoking, and clubfoot: an indication of a gene-environment interaction. Am J Epidemiol 152: 658-665. doi: 10.1093/aje/152.7.658
|
| [117] |
Gurnett CA, Alaee F, Kruse LM, et al. (2008) Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am J Hum Genet 83: 616-622. doi: 10.1016/j.ajhg.2008.10.004
|
| [118] |
Alvarado DM, McCall K, Aferol H, et al. (2011) Pitx1 haploinsufficiency causes clubfoot in humans and a clubfoot-like phenotype in mice. Hum Mol Genet 20: 3943-3952. doi: 10.1093/hmg/ddr313
|
| [119] |
Yong BC, Xun FX, Zhao LJ, et al. (2016) A systematic review of association studies of common variants associated with idiopathic congenital talipes equinovarus (ICTEV) in humans in the past 30 years. Springerplus 5: 896-016-2353-8. eCollection 2016. doi: 10.1186/s40064-016-2353-8
|
| [120] |
Rodriguez-Esteban C, Tsukui T, Yonei S, et al. (1999) The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature 398: 814-818. doi: 10.1038/19769
|
| [121] | Alvarado DM, Aferol H, McCall K, et al. (2010) Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am J Hum Genet 87: 154-160. |
| [122] | Lu W, Bacino CA, Richards BS, et al. (2012) Studies of TBX4 and chromosome 17q23.1q23.2: an uncommon cause of nonsyndromic clubfoot. Am J Med Genet A 158A: 1620-1627. |
| [123] |
Alnemri ES, Livingston DJ, Nicholson DW, et al. (1996) Human ICE/CED-3 protease nomenclature. Cell 87: 171. doi: 10.1016/S0092-8674(00)81334-3
|
| [124] |
Heck AL, Bray MS, Scott A, et al. (2005) Variation in CASP10 gene is associated with idiopathic talipes equinovarus. J Pediatr Orthop 25: 598-602. doi: 10.1097/01.bpo.0000173248.96936.90
|
| [125] |
Ester AR, Tyerman G, Wise CA, et al. (2007) Apoptotic gene analysis in idiopathic talipes equinovarus (clubfoot). Clin Orthop Relat Res 462: 32-37. doi: 10.1097/BLO.0b013e318073c2d9
|
| [126] |
Daher S, Guimaraes AJ, Mattar R, et al. (2008) Bcl-2 and Bax expressions in pre-term, term and post-term placentas. Am J Reprod Immunol 60: 172-178. doi: 10.1111/j.1600-0897.2008.00609.x
|
| [127] |
Peebles DM (2004) Fetal consequences of chronic substrate deprivation. Semin Fetal Neonatal Med 9: 379-386. doi: 10.1016/j.siny.2004.03.008
|
| [128] |
Sundberg K, Bang J, Smidt-Jensen S, et al. (1997) Randomised study of risk of fetal loss related to early amniocentesis versus chorionic villus sampling. Lancet 350: 697-703. doi: 10.1016/S0140-6736(97)02449-5
|
| [129] |
Cederholm M, Haglund B, Axelsson O (2005) Infant morbidity following amniocentesis and chorionic villus sampling for prenatal karyotyping. BJOG 112: 394-402. doi: 10.1111/j.1471-0528.2005.00413.x
|
| [130] |
Mark M, Rijli FM, Chambon P (1997) Homeobox genes in embryogenesis and pathogenesis. Pediatr Res 42: 421-429. doi: 10.1203/00006450-199710000-00001
|
| [131] |
McGinnis W, Krumlauf R (1992) Homeobox genes and axial patterning. Cell 68: 283-302. doi: 10.1016/0092-8674(92)90471-N
|
| [132] |
Dobbs MB, Gurnett CA, Pierce B, et al. (2006) HOXD10 M319K mutation in a family with isolated congenital vertical talus. J Orthop Res 24: 448-453. doi: 10.1002/jor.20052
|
| [133] |
Shrimpton AE, Levinsohn EM, Yozawitz JM, et al. (2004) A HOX gene mutation in a family with isolated congenital vertical talus and Charcot-Marie-Tooth disease. Am J Hum Genet 75: 92-96. doi: 10.1086/422015
|
| [134] | Weymouth KS, Blanton SH, Bamshad MJ, et al. (2011) Variants in genes that encode muscle contractile proteins influence risk for isolated clubfoot. Am J Med Genet A 155A: 2170-2179. |
| [135] |
McKillop DF, Geeves MA (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65: 693-701. doi: 10.1016/S0006-3495(93)81110-X
|
| [136] | Gordon AM, Homsher E, Regnier M (2000) Regulation of contraction in striated muscle. Physiol Rev 80: 853-924. |
| [137] |
Weymouth KS, Blanton SH, Powell T, et al. (2016) Functional Assessment of Clubfoot Associated HOXA9, TPM1, and TPM2 Variants Suggests a Potential Gene Regulation Mechanism. Clin Orthop Relat Res 474: 1726-1735. doi: 10.1007/s11999-016-4788-1
|
| [138] |
Castaneda C, Nalley K, Mannion C, et al. (2015) Clinical decision support systems for improving diagnostic accuracy and achieving precision medicine. J Clin Bioinforma 5: 4-015-0019-3. eCollection 2015. doi: 10.1186/s13336-015-0019-3
|
| [139] |
Rehm HL (2013) Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet 14: 295-300. doi: 10.1038/nrg3463
|
| [140] |
Richards S, Aziz N, Bale S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405-424. doi: 10.1038/gim.2015.30
|
| [141] |
Green RC, Berg JS, Grody WW, et al. (2013) ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 15: 565-574. doi: 10.1038/gim.2013.73
|


Figures(4)

Neha Sinha, Mark A. Seeley, Daniel S. Horwitz, Hemil Maniar, Andrea H. Seeley. Pediatric Orthogenomics: The Latest Trends and Controversies[J]. AIMS Medical Science, 2017, 4(2): 192-216. doi: 10.3934/medsci.2017.2.192







 DownLoad:
DownLoad: