Identifying key proteins based on protein-protein interaction networks has emerged as a prominent area of research in bioinformatics. However, current methods exhibit certain limitations, such as the omission of subcellular localization information and the disregard for the impact of topological structure noise on the reliability of key protein identification. Moreover, the influence of proteins outside a complex but interacting with proteins inside the complex on complex participation tends to be overlooked. Addressing these shortcomings, this paper presents a novel method for key protein identification that integrates protein complex information with multiple biological features. This approach offers a comprehensive evaluation of protein importance by considering subcellular localization centrality, topological centrality weighted by gene ontology (GO) similarity and complex participation centrality. Experimental results, including traditional statistical metrics, jackknife methodology metric and key protein overlap or difference, demonstrate that the proposed method not only achieves higher accuracy in identifying key proteins compared to nine classical methods but also exhibits robustness across diverse protein-protein interaction networks.
Citation: Yongyin Han, Maolin Liu, Zhixiao Wang. Key protein identification by integrating protein complex information and multi-biological features[J]. Mathematical Biosciences and Engineering, 2023, 20(10): 18191-18206. doi: 10.3934/mbe.2023808
Identifying key proteins based on protein-protein interaction networks has emerged as a prominent area of research in bioinformatics. However, current methods exhibit certain limitations, such as the omission of subcellular localization information and the disregard for the impact of topological structure noise on the reliability of key protein identification. Moreover, the influence of proteins outside a complex but interacting with proteins inside the complex on complex participation tends to be overlooked. Addressing these shortcomings, this paper presents a novel method for key protein identification that integrates protein complex information with multiple biological features. This approach offers a comprehensive evaluation of protein importance by considering subcellular localization centrality, topological centrality weighted by gene ontology (GO) similarity and complex participation centrality. Experimental results, including traditional statistical metrics, jackknife methodology metric and key protein overlap or difference, demonstrate that the proposed method not only achieves higher accuracy in identifying key proteins compared to nine classical methods but also exhibits robustness across diverse protein-protein interaction networks.

| [1] |
L. yan Wang, Z. Zhang, Y. Li, Y. Wan, B. Xing, Integrated bioinformatic analysis of rna binding proteins in hepatocellular carcinoma, Aging (Albany NY), 13 (2020), 2480–2505. https://doi.org/10.18632/aging.202281 doi: 10.18632/aging.202281

|
| [2] |
X. Wang, J. Zhao, Targeted cancer therapy based on acetylation and deacetylation of key proteins involved in double-strand break repair, Cancer Manag. Res., (2022), 259–271. https://doi.org/10.2147/CMAR.S346052 doi: 10.2147/CMAR.S346052
|
| [3] |
Y. Yue, C. Ye, P.-Y. Peng, H.-X. Zhai, I. Ahmad, C. Xia, et al., A deep learning framework for identifying essential proteins based on multiple biological information, BMC Bioinform., 23 (2022), 318. https://doi.org/10.1186/s12859-022-04868-8 doi: 10.1186/s12859-022-04868-8
|
| [4] |
Y. Liu, W. Chen, Z. He, Essential protein recognition via community significance, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 18 (2021), 2788–2794. https://doi.org/10.1109/TCBB.2021.3102018 doi: 10.1109/TCBB.2021.3102018
|
| [5] |
L. Shen, J. Zhang, F. Wang, K. Liu, Predicting essential proteins based on integration of local fuzzy fractal dimension and subcellular location information, Genes, 13 (2022), 173. https://doi.org/10.3390/genes13020173 doi: 10.3390/genes13020173
|
| [6] |
X.-J. Lei, Y. Gao, L. Guo, Mining protein complexes based on topology potential weight in dynamic protein-protein interaction networks, Acta Electon. Sin., 46 (2018), 145. https://doi.org/10.3969/j.issn.0372-2112.2018.01.020 doi: 10.3969/j.issn.0372-2112.2018.01.020
|
| [7] |
T. Tang, X. Zhang, Y. Liu, H. Peng, B. Zheng, et al., Machine learning on protein–protein interaction prediction: models, challenges and trends, Brief. Bioinform., 24 (2023), bbad076. https://doi.org/10.1093/bib/bbad076 doi: 10.1093/bib/bbad076
|
| [8] |
M. Li, H. Zhang, J.-x. Wang, Y. Pan, A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data, BMC Syst. Biol., 6 (2012), 1–9. https://doi.org/10.1186/1752-0509-6-15 doi: 10.1186/1752-0509-6-15
|
| [9] |
W. Peng, J. Wang, Y. Cheng, Y. Lu, F. Wu, Y. Pan, Udonc: An algorithm for identifying essential proteins based on protein domains and protein-protein interaction networks, IEEE/ACM Transact. Comput. Biol. Bioinform., 12 (2014), 276–288. https://doi.org/10.1109/TCBB.2014.2338317 doi: 10.1109/TCBB.2014.2338317
|
| [10] |
X. Shang, Y. Wang, B. Chen, Identifying essential proteins based on dynamic protein-protein interaction networks and rna-seq datasets, Sci. China Inform. Sci., 59 (2016), 1–11. https://doi.org/10.1007/s11432-016-5583-z doi: 10.1007/s11432-016-5583-z
|
| [11] |
M. LI, X.-t. WANG, H.-m. LUO, X.-m. MENG, J.-x. WANG, Progress on random walk and its application in network biology, Acta Electon. Sin., 46 (2018), 2035. https://doi.org/10.3969/j.issn.0372-2112.2018.08.033 doi: 10.3969/j.issn.0372-2112.2018.08.033
|
| [12] |
M. Li, Y. Lu, Z. Niu, F.-X. Wu, United complex centrality for identification of essential proteins from ppi networks, IEEE/ACM Transact. Comput. Biol. Bioinform., 14 (2015), 370–380. https://doi.org/10.1109/TCBB.2015.2394487 doi: 10.1109/TCBB.2015.2394487
|
| [13] |
J. Zhong, C. Tang, W. Peng, M. Xie, Y. Sun, Q. Tang, et al., A novel essential protein identification method based on ppi networks and gene expression data, BMC Bioinform., 22 (2021), 1–21. https://doi.org/10.1186/s12859-021-04175-8 doi: 10.1186/s12859-021-04175-8
|
| [14] |
C. Qin, Y. Sun, Y. Dong, A new method for identifying essential proteins based on network topology properties and protein complexes, PloS One, 11 (2016), e0161042. https://doi.org/10.1371/journal.pone.0161042 doi: 10.1371/journal.pone.0161042
|
| [15] |
G. Yu, G. Fu, J. Wang and H. Zhu, Predicting protein function via semantic integration of multiple networks, IEEE/ACM Transact. Comput. Biol. Bioinform., 13 (2015), 220–232. https://doi.org/10.1109/TCBB.2015.2459713 doi: 10.1109/TCBB.2015.2459713
|
| [16] |
J. Luo, Y. Qi, Identification of essential proteins based on a new combination of local interaction density and protein complexes, PloS One, 10 (2015), e0131418. https://doi.org/10.1371/journal.pone.0131418 doi: 10.1371/journal.pone.0131418
|
| [17] |
X. Yang, Z. Niu, Y. Liu, B. Song, W. Lu, L. Zeng, et al., Modality-dta: Multimodality fusion strategy for drug–target affinity prediction, IEEE/ACM Transact. Comput. Biol. Bioinform., 20 (2022), 1200–1210. https://doi.org/10.1109/TCBB.2022.3205282 doi: 10.1109/TCBB.2022.3205282
|
| [18] |
W. Zhang, J. Xu, Y. Li, X. Zou, Detecting essential proteins based on network topology, gene expression data, and gene ontology information, IEEE/ACM Transact. Comput. Biol. Bioinform., 15 (2016), 109–116. https://doi.org/10.1109/TCBB.2016.2615931 doi: 10.1109/TCBB.2016.2615931
|
| [19] |
B. Chen, W. Fan, J. Liu, F.-X. Wu, Identifying protein complexes and functional modules—from static ppi networks to dynamic ppi networks, Brief. Bioinform., 15 (2014), 177–194. https://doi.org/10.1093/bib/bbt039 doi: 10.1093/bib/bbt039
|
| [20] |
R. R. Vallabhajosyula, D. Chakravarti, S. Lutfeali, A. Ray, A. Raval, Identifying hubs in protein interaction networks, PloS One, 4 (2009), e5344. https://doi.org/10.1371/journal.pone.0005344 doi: 10.1371/journal.pone.0005344
|
| [21] |
M. P. Joy, A. Brock, D. E. Ingber, S. Huang, High-betweenness proteins in the yeast protein interaction network, J. Biomed. Biotechnol., 2005 (2005), 96. https://doi.org/10.1155/JBB.2005.96 doi: 10.1155/JBB.2005.96
|
| [22] |
E. Estrada, J. A. Rodriguez-Velazquez, Subgraph centrality in complex networks, Phys. Rev. E, 71 (2005), 056103. https://doi.org/10.1103/PhysRevE.71.056103 doi: 10.1103/PhysRevE.71.056103
|
| [23] |
J. Wang, M. Li, H. Wang, Y. Pan, Identification of essential proteins based on edge clustering coefficient, IEEE/ACM Transact. Comput. Biol. Bioinform., 9 (2011), 1070–1080. https://doi.org/10.1109/TCBB.2011.147 doi: 10.1109/TCBB.2011.147
|
| [24] |
P. Lu, J. Yu, A mixed clustering coefficient centrality for identifying essential proteins, Int. J. Modern Phys. B, 34 (2020), 2050090. https://doi.org/10.1142/S0217979220500897 doi: 10.1142/S0217979220500897
|
| [25] |
I. Xenarios, L. Salwinski, X. J. Duan, P. Higney, S.-M. Kim, D. Eisenberg, Dip, the database of interacting proteins: a research tool for studying cellular networks of protein interactions, Nucleic Acids Res., 30 (2002), 303–305. https://doi.org/10.1093/nar/28.1.289 doi: 10.1093/nar/28.1.289
|
| [26] |
N. J. Krogan, G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, et al., Global landscape of protein complexes in the yeast saccharomyces cerevisiae, Nature, 440 (2006), 637–643. https://doi.org/10.1038/nature04670 doi: 10.1038/nature04670
|
| [27] |
U. Güldener, M. Münsterkötter, M. Oesterheld, P. Pagel, A. Ruepp, H.-W. Mewes, et al., Mpact: The mips protein interaction resource on yeast, Nucleic Acids Res., 34 (2006), D436–D441. https://doi.org/10.1093/nar/gkj003 doi: 10.1093/nar/gkj003
|
| [28] |
G. O. Consortium, Gene ontology annotations and resources, Nucleic Acids Res., 41 (2012), D530–D535. https://doi.org/10.1093/nar/gks1050 doi: 10.1093/nar/gks1050
|
| [29] |
J. X. Binder, S. Pletscher-Frankild, K. Tsafou, C. Stolte, S. I. O'Donoghue, R. Schneider, et al., Compartments: Unification and visualization of protein subcellular localization evidence, Database, 2014. https://doi.org/10.1093/database/bau012 doi: 10.1093/database/bau012
|
| [30] |
R. Zhang, Y. Lin, Deg 5.0, a database of essential genes in both prokaryotes and eukaryotes, Nucleic Acids Res., 37 (2009), D455–D458. https://doi.org/10.1093/nar/gkn858 doi: 10.1093/nar/gkn858
|
| [31] |
H.-W. Mewes, C. Amid, R. Arnold, D. Frishman, U. Güldener, G. Mannhaupt, et al., Mips: Analysis and annotation of proteins from whole genomes, Nucleic Acids Res., 32 (2004), D41–D44. https://doi.org/10.1093/nar/gkh092 doi: 10.1093/nar/gkh092
|
| [32] |
J. M. Cherry, C. Adler, C. Ball, S. A. Chervitz, S. S. Dwight, E. T. Hester, et al., Sgd: Saccharomyces genome database, Nucleic Acids Res., 26 (1998), 73–79. https://doi.org/10.1093/nar/26.1.73 doi: 10.1093/nar/26.1.73
|
| [33] |
E. A. Winzeler, D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, et al., Functional characterization of the s. cerevisiae genome by gene deletion and parallel analysis, Science, 285 (1999), 901–906. https://doi.org/10.1126/science.285.5429.90 doi: 10.1126/science.285.5429.90
|
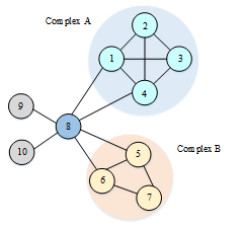
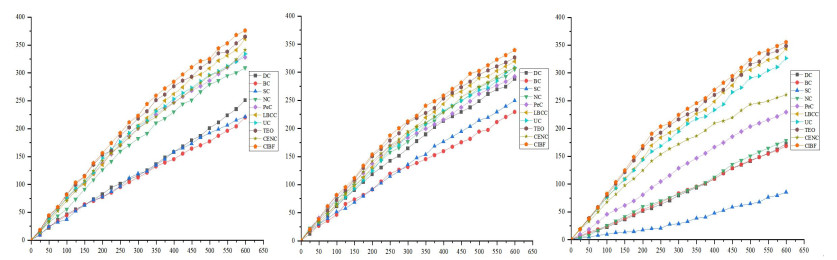
Figures(2) / Tables(15)

Yongyin Han, Maolin Liu, Zhixiao Wang. Key protein identification by integrating protein complex information and multi-biological features[J]. Mathematical Biosciences and Engineering, 2023, 20(10): 18191-18206. doi: 10.3934/mbe.2023808







 DownLoad:
DownLoad: