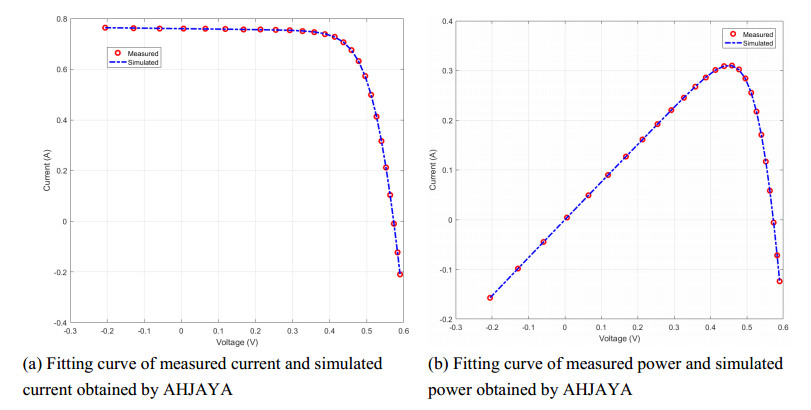
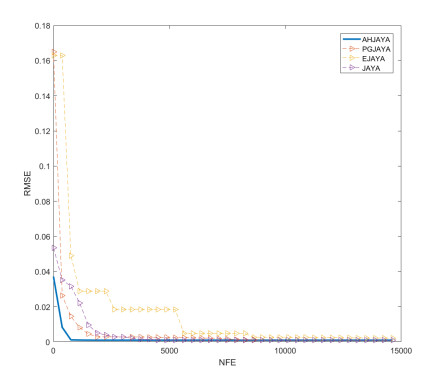
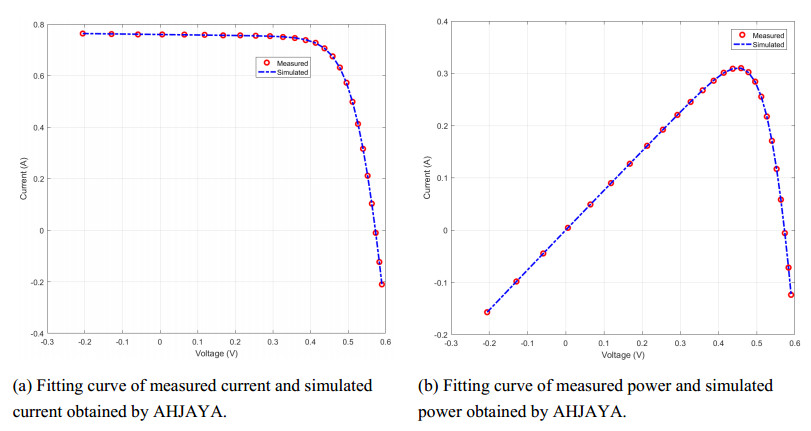
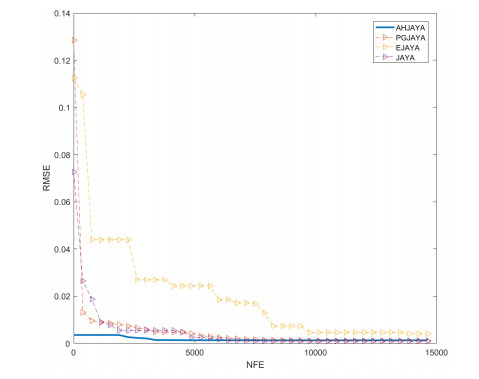
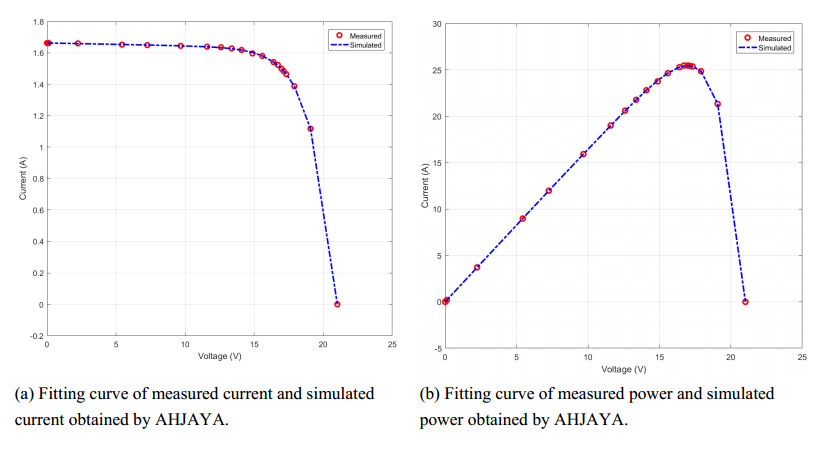
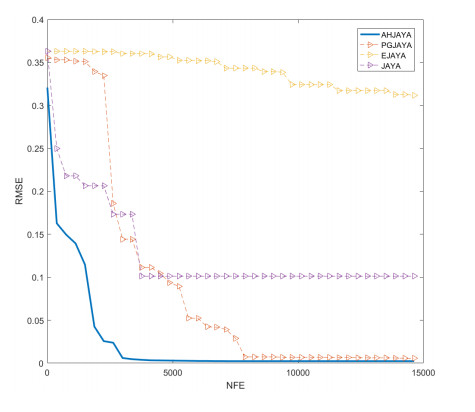
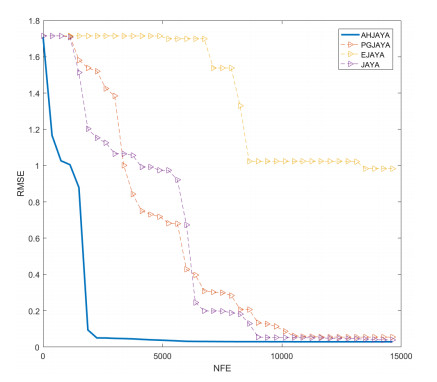
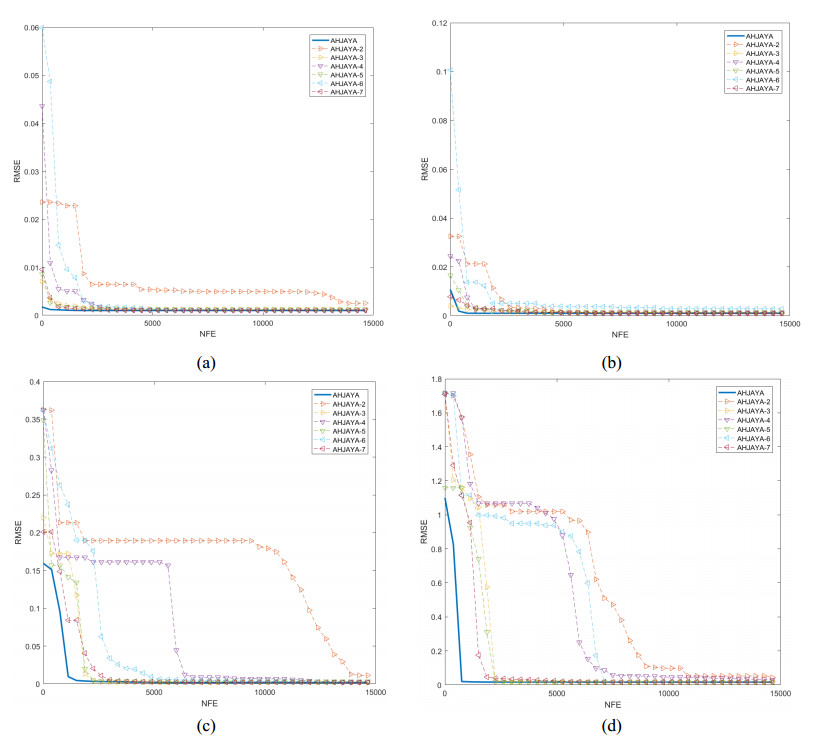
In order to have the highest efficiency in real-life photovoltaic power generation systems, how to model, optimize and control photovoltaic systems has become a challenge. The photovoltaic power generation systems are dominated by photovoltaic models, and its performance depends on its unknown parameters. However, the modeling equation of the photovoltaic model is nonlinear, leading to the difficulty in parameter extraction. To extract the parameters of the photovoltaic model more accurately and efficiently, a chaotic self-adaptive JAYA algorithm, called AHJAYA, was proposed, where various improvement strategies are introduced. First, self-adaptive coefficients are introduced to change the priority of information from the best search agent and the worst search agent. Second, by combining the linear population reduction strategy with the chaotic opposition-based learning strategy, the convergence speed of the algorithm is improved as well as avoid falling into local optimum. To verify the performance of the AHJAYA, four photovoltaic models are selected. The experimental results prove that the proposed AHJAYA has superior performance and strong competitiveness.
Citation: Juan Zhao, Yujun Zhang, Shuijia Li, Yufei Wang, Yuxin Yan, Zhengming Gao. A chaotic self-adaptive JAYA algorithm for parameter extraction of photovoltaic models[J]. Mathematical Biosciences and Engineering, 2022, 19(6): 5638-5670. doi: 10.3934/mbe.2022264
In order to have the highest efficiency in real-life photovoltaic power generation systems, how to model, optimize and control photovoltaic systems has become a challenge. The photovoltaic power generation systems are dominated by photovoltaic models, and its performance depends on its unknown parameters. However, the modeling equation of the photovoltaic model is nonlinear, leading to the difficulty in parameter extraction. To extract the parameters of the photovoltaic model more accurately and efficiently, a chaotic self-adaptive JAYA algorithm, called AHJAYA, was proposed, where various improvement strategies are introduced. First, self-adaptive coefficients are introduced to change the priority of information from the best search agent and the worst search agent. Second, by combining the linear population reduction strategy with the chaotic opposition-based learning strategy, the convergence speed of the algorithm is improved as well as avoid falling into local optimum. To verify the performance of the AHJAYA, four photovoltaic models are selected. The experimental results prove that the proposed AHJAYA has superior performance and strong competitiveness.

| [1] |
S. Li, W. Gong, X. Yan, C. Hu, D. Bai, L. Wang, Parameter estimation of photovoltaic models with memetic adaptive differential evolution, Sol. Energy, 190 (2019), 465–474. https://doi.org/10.1016/j.solener.2019.08.022 doi: 10.1016/j.solener.2019.08.022

|
| [2] | Z. Liao, Q. Gu, S. Li, Z. Hu, B. Ning, An improved differential evolution to extract photovoltaic cell parameters, IEEE Access, 8 (2020), 177838–177850. http://doi.org/10.1109/ACCESS.2020.3024975 |
| [3] |
S. Li, Q. Gu, W. Gong, B. Ning, An enhanced adaptive differential evolution algorithm for parameter extraction of photovoltaic models, Energy Convers. Manage., 205 (2020), 112443. https://doi.org/10.1016/j.enconman.2019.112443 doi: 10.1016/j.enconman.2019.112443
|
| [4] |
Z. Liao, Z. Chen, S. Li, Parameters extraction of photovoltaic models using triple-phase teaching-learning-based optimization, IEEE Access, 8 (2020), 69937–69952. https://doi.org/10.1109/ACCESS.2020.2984728 doi: 10.1109/ACCESS.2020.2984728
|
| [5] |
H. M. Ridha, H. Hizam, C. Gomes, A. A. Heidari, H. Chen, M. Ahmadipour, et al., Parameters extraction of three diode photovoltaic models using boosted LSHADE algorithm and Newton Raphson method, Energy, 224 (2021), 120136. https://doi.org/10.1016/j.energy.2021.120136 doi: 10.1016/j.energy.2021.120136
|
| [6] |
S. Li, W. Gong, Q. Gu, A comprehensive survey on meta-heuristic algorithms for parameter extraction of photovoltaic models, Renewable Sustainable Energy Rev., 141 (2021), 110828. https://doi.org/10.1016/j.rser.2021.110828 doi: 10.1016/j.rser.2021.110828
|
| [7] |
M. Abd Elaziz, D. Oliva, Parameter estimation of solar cells diode models by an improved opposition-based whale optimization algorithm, Energy Convers. Manage., 171 (2018), 1843–1859. https://doi.org/10.1016/j.enconman.2018.05.062 doi: 10.1016/j.enconman.2018.05.062
|
| [8] |
J. Liang, K. Qiao, M. Yuan, K. Yu, B. Qu, S. Ge, et al., Evolutionary multi-task optimization for parameters extraction of photovoltaic models, Energy Convers. Manage., 207 (2020), 112509. https://doi.org/10.1016/j.enconman.2020.112509 doi: 10.1016/j.enconman.2020.112509
|
| [9] |
A. Askarzadeh, A. Rezazadeh, Parameter identification for solar cell models using harmony search-based algorithms, Sol. Energy, 86 (2012), 3241–3249. https://doi.org/10.1016/j.solener.2012.08.018 doi: 10.1016/j.solener.2012.08.018
|
| [10] |
T. Kang, J. Yao, M. Jin, S. Yang, T. Duong, A novel improved cuckoo search algorithm for parameter estimation of photovoltaic (PV) models, Energies, 11 (2018), 1–31. https://doi.org/10.3390/en11051060 doi: 10.3390/en11051060
|
| [11] |
M. R. AlRashidi, M. F. AlHajri, K. M. El-Naggar, A. K. Al-Othman, A new estimation approach for determining the Ⅰ–Ⅴ characteristics of solar cells, Sol. Energy, 85 (2011), 1543–1550. https://doi.org/10.1016/j.solener.2011.04.013 doi: 10.1016/j.solener.2011.04.013
|
| [12] |
A. Askarzadeh, A. Rezazadeh, Artificial bee swarm optimization algorithm for parameters identification of solar cell models, Appl. Energy, 102 (2013), 943–949. https://doi.org/10.1016/j.apenergy.2012.09.052 doi: 10.1016/j.apenergy.2012.09.052
|
| [13] |
R. Ben Messaoud, Extraction of uncertain parameters of single-diode model of a photovoltaic panel using simulated annealing optimization, Energy Rep., 6 (2020), 350–357. https://doi.org/10.1016/j.egyr.2020.01.016 doi: 10.1016/j.egyr.2020.01.016
|
| [14] |
S. Li, W. Gong, L. Wang, X. Yan, C. Hu, A hybrid adaptive teaching–learning-based optimization and differential evolution for parameter identification of photovoltaic models, Energy Convers. Manage., 225 (2020), 113474. https://doi.org/10.1016/j.enconman.2020.113474 doi: 10.1016/j.enconman.2020.113474
|
| [15] |
K. G. K. Harish, Modeling of solar cell under different conditions by Ant Lion Optimizer with LambertW function, Appl. Soft Comput., 71 (2018), 141–151. https://doi.org/10.1016/j.asoc.2018.06.025 doi: 10.1016/j.asoc.2018.06.025
|
| [16] |
H. M. Ridha, H. Hizam, S. Mirjalili, M. L. Othman, M. E. Ya'acob, L. Abualigah, A novel theoretical and practical methodology for extracting the parameters of the single and double diode photovoltaic models, IEEE Access, 10 (2022), 11110–11137. https://doi.org/10.1109/ACCESS.2022.3142779 doi: 10.1109/ACCESS.2022.3142779
|
| [17] |
A. A. Al-Shamma'a, H. O. Omotoso, F. A. Alturki, H. M. H. Farh, A. Alkuhayli, K. Alsharabi, et al., Parameter estimation of photovoltaic cell/modules using bonobo optimizer, Energies, 15 (2022), 140. https://doi.org/10.3390/en15010140 doi: 10.3390/en15010140
|
| [18] |
W. Zhou, P. Wang, A. A. Heidari, X. Zhao, H. Turabieh, M. Mafarja, et al., Metaphor-free dynamic spherical evolution for parameter estimation of photovoltaic modules, Energy Rep., 7 (2021), 5175–5202. https://doi.org/10.1016/j.egyr.2021.07.041 doi: 10.1016/j.egyr.2021.07.041
|
| [19] |
A. Farah, A. Belazi, F. Benabdallah, A. Almalaq, M. Chtourou, M. A. Abido, Parameter extraction of photovoltaic models using a comprehensive learning Rao-1 algorithm, Energy Convers. Manage., 252 (2022), 115057. https://doi.org/10.1016/j.enconman.2021.115057 doi: 10.1016/j.enconman.2021.115057
|
| [20] |
J. Luo, J. Zhou, X. Jiang, A modification of the imperialist competitive algorithm with hybrid methods for constrained optimization problems, IEEE Access, 9 (2021), 161745–161760. https://doi.org/10.1109/ACCESS.2021.3133579 doi: 10.1109/ACCESS.2021.3133579
|
| [21] |
M. A. E. Sattar, A. Al Sumaiti, H. Ali, A. A. Z. Diab, Marine predators algorithm for parameters estimation of photovoltaic modules considering various weather conditions, Neural Comput. Appl., 33 (2021), 11799–11819. https://doi.org/10.1007/s00521-021-05822-0 doi: 10.1007/s00521-021-05822-0
|
| [22] |
S. Jiao, G. Chong, C. Huang, H. Hu, M. Wang, A. A. Heidari, et al., Orthogonally adapted Harris hawks optimization for parameter estimation of photovoltaic models, Energy, 203 (2020), 117804. https://doi.org/10.1016/j.energy.2020.117804 doi: 10.1016/j.energy.2020.117804
|
| [23] |
Y. Yu, K. Wang, T. Zhang, Y. Wang, C. Peng, S. Gao, A population diversity-controlled differential evolution for parameter estimation of solar photovoltaic models, Sustainable Energy Technol. Assess., 51 (2022), 101938. https://doi.org/10.1016/j.seta.2021.101938 doi: 10.1016/j.seta.2021.101938
|
| [24] |
S. Gao, K. Wang, S. Tao, T. Jin, H. Dai, J. Cheng, A state-of-the-art differential evolution algorithm for parameter estimation of solar photovoltaic models, Energy Convers. Manage., 230 (2021), 113784. https://doi.org/10.1016/j.enconman.2020.113784 doi: 10.1016/j.enconman.2020.113784
|
| [25] | R. V. Rao, Jaya: A simple and new optimization algorithm for solving constrained and unconstrained optimization problems, Int. J. Ind. Eng. Comput., 7 (2016), 19–34. http://dx.doi.org/10.5267/j.ijiec.2015.8.004 |
| [26] |
Y. Zhang, Z. Jin, Comprehensive learning Jaya algorithm for engineering design optimization problems, J. Intell. Manuf., 2021 (2021). https://doi.org/10.1007/s10845-020-01723-6 doi: 10.1007/s10845-020-01723-6
|
| [27] |
Y. Zhang, A. Chi, S. Mirjalili, Enhanced Jaya algorithm: A simple but efficient optimization method for constrained engineering design problems, Knowl. Based Syst., 233 (2021), 107555. https://doi.org/10.1016/j.knosys.2021.107555 doi: 10.1016/j.knosys.2021.107555
|
| [28] |
M. Afifi, H. Rezk, M. Ibrahim, M. El-Nemr, Multi-objective optimization of switched reluctance machine design using jaya algorithm (MO-Jaya), Mathematics, 9 (2021), 1107. https://doi.org/10.3390/math9101107 doi: 10.3390/math9101107
|
| [29] |
S. Basak, B. Bhattacharyya, B. Dey, Combined economic emission dispatch on dynamic systems using hybrid CSA-JAYA Algorithm, Int. J. Syst. Assur. Eng. Manage., 2022 (2022). https://doi.org/10.1007/s13198-022-01635-z doi: 10.1007/s13198-022-01635-z
|
| [30] |
D. Saadaoui, M. Elyaqouti, K. Assalaou, D. B. hmamou, S. Lidaighbi, Multiple learning JAYA algorithm for parameters identifying of photovoltaic models, Mater. Today Proc., 52 (2022), 108–123. https://doi.org/10.1016/j.matpr.2021.11.106 doi: 10.1016/j.matpr.2021.11.106
|
| [31] |
M. F. Tefek, M. Arslan, Highway accident number estimation in Turkey with Jaya algorithm, Neural Comput. Appl., 34 (2022), 5367–5381. https://doi.org/10.1007/s00521-022-06952-9 doi: 10.1007/s00521-022-06952-9
|
| [32] |
J. Gholami, M. R. Kamankesh, S. Mohammadi, E. Hosseinkhani, S. Abdi, Powerful enhanced Jaya algorithm for efficiently optimizing numerical and engineering problems, Soft Comput., 2022 (2022). https://doi.org/10.1007/s00500-022-06909-z doi: 10.1007/s00500-022-06909-z
|
| [33] |
X. Jian, Y. Cao, A chaotic second order oscillation JAYA Algorithm for parameter extraction of photovoltaic models, Photonics, 9 (2022). https://doi.org/10.3390/photonics9030131 doi: 10.3390/photonics9030131
|
| [34] |
S. Belagoune, N. Bali, K. Atif, H. Labdelaoui, A discrete chaotic Jaya algorithm for optimal preventive maintenance scheduling of power systems generators, Appl. Soft Comput., 119 (2022), 108608. https://doi.org/10.1016/j.asoc.2022.108608 doi: 10.1016/j.asoc.2022.108608
|
| [35] |
A. Aleti, I. Moser, A systematic literature review of adaptive parameter control methods for evolutionary algorithms, Assoc. Comput. Mach., 49 (2017), 1–35. https://doi.org/10.1145/2996355 doi: 10.1145/2996355
|
| [36] |
Z. Lei, S. Gao, S. Gupta, J. Cheng, G. Yang, An aggregative learning gravitational search algorithm with self-adaptive gravitational constants, Exp. Syst. Appl., 152 (2020), 113396. https://doi.org/10.1016/j.eswa.2020.113396 doi: 10.1016/j.eswa.2020.113396
|
| [37] | R. Tanabe, A. S. Fukunaga, Improving the search performance of SHADE using linear population size reduction, in 2014 IEEE Congress on Evolutionary Computation (CEC), (2014), 1658–1665. https://doi.org/10.1109/CEC.2014.6900380 |
| [38] | H. Yang, S. Gao, R. L. Wang, Y. Todo, A ladder spherical evolution search algorithm, IEICE Trans. Inf. Syst., 104 (2021), 461–464. http://doi.org/10.1587/transinf.2020EDL8102 |
| [39] |
X. Yu, X. Wu, W. Luo, Parameter identification of photovoltaic models by hybrid adaptive JAYA Algorithm, Mathematics, 10 (2022), 183. https://doi.org/10.3390/math10020183 doi: 10.3390/math10020183
|
| [40] |
Y. J. Zhang, Y. X. Yan, J. Zhao, Z. M. Gao, AOAAO: The hybrid algorithm of arithmetic optimization algorithm with aquila optimizer, IEEE Access, 10 (2022), 10907–10933. https://doi.org/10.1109/ACCESS.2022.3144431 doi: 10.1109/ACCESS.2022.3144431
|
| [41] |
J. Zhao, Z.-M. Gao, The chaotic slime mould algorithm with chebyshev map, in 2nd International Conference on Artificial Intelligence and Computer Science, 1631 (2020), 012071. https://doi.org/10.1088/1742-6596/1631/1/012071 doi: 10.1088/1742-6596/1631/1/012071
|
| [42] |
K. Yu, B. Qu, C. Yue, S. Ge, X. Chen, J. Liang, A performance-guided JAYA algorithm for parameters identification of photovoltaic cell and module, Appl. Energy, 237 (2019), 241–257. https://doi.org/10.1016/j.apenergy.2019.01.008 doi: 10.1016/j.apenergy.2019.01.008
|
| [43] |
Z. Yan, S. Li, W. Gong, An adaptive differential evolution with decomposition for photovoltaic parameter extraction, Math. Biosci. Eng., 18 (2021), 7363–7388. https://doi.org/10.1016/j.apenergy.2019.01.008 doi: 10.1016/j.apenergy.2019.01.008
|
| [44] |
G. Xiong, J. Zhang, X. Yuan, D. Shi, Y. He, G. Yao, Parameter extraction of solar photovoltaic models by means of a hybrid differential evolution with whale optimization algorithm, Sol. Energy, 176 (2018), 742–761. https://doi.org/10.1016/j.solener.2018.10.050 doi: 10.1016/j.solener.2018.10.050
|
| [45] |
S. Li, W. Gong, X. Yan, C. Hu, D. Bai, L. Wang, et al., Parameter extraction of photovoltaic models using an improved teaching-learning-based optimization, Energy Convers. Manage., 186 (2019), 293–305. https://doi.org/10.1016/j.enconman.2019.02.048 doi: 10.1016/j.enconman.2019.02.048
|
| [46] |
X. Chen, K. Yu, Hybridizing cuckoo search algorithm with biogeography-based optimization for estimating photovoltaic model parameters, Sol. Energy, 180 (2019), 192–206. https://doi.org/10.1016/j.solener.2019.01.025 doi: 10.1016/j.solener.2019.01.025
|
| [47] |
L. M. P. Deotti, J. L. R. Pereira, I. C. Silva Júnior, Parameter extraction of photovoltaic models using an enhanced Lévy flight bat algorithm, Energy Convers. Manage., 221 (2020), 113114. https://doi.org/10.1016/j.enconman.2020.113114 doi: 10.1016/j.enconman.2020.113114
|
| [48] |
G. Xiong, J. Zhang, D. Shi, L. Zhu, X. Yuan, Parameter extraction of solar photovoltaic models with an either-or teaching learning based algorithm, Energy Convers. Manage., 224 (2020), 113395. https://doi.org/10.1016/j.enconman.2020.113395 doi: 10.1016/j.enconman.2020.113395
|
| [49] |
X. Yang, W. Gong, Opposition-based JAYA with population reduction for parameter estimation of photovoltaic solar cells and modules, Appl. Soft Comput., 104 (2021), 107218. https://doi.org/10.1016/j.asoc.2021.107218 doi: 10.1016/j.asoc.2021.107218
|
| [50] |
K. M. Sallam, M. A. Hossain, R. K. Chakrabortty, M. J. Ryan, An improved gaining-sharing knowledge algorithm for parameter extraction of photovoltaic models, Energy Convers. Manage., 237 (2021), 114030. https://doi.org/10.1016/j.enconman.2021.114030 doi: 10.1016/j.enconman.2021.114030
|
| [51] |
Z. Hu, W. Gong, S. Li, Reinforcement learning-based differential evolution for parameters extraction of photovoltaic models, Energy Rep., 7 (2021), 916–928. https://doi.org/10.1016/j.egyr.2021.01.096 doi: 10.1016/j.egyr.2021.01.096
|
| [52] |
K. Yu, J. J. Liang, B. Y. Qu, Z. Cheng, H. Wang, Multiple learning backtracking search algorithm for estimating parameters of photovoltaic models, Appl. Energy, 226 (2018), 408–422. https://doi.org/10.1016/j.apenergy.2018.06.010 doi: 10.1016/j.apenergy.2018.06.010
|
| [53] |
N. Pourmousa, S. M. Ebrahimi, M. Malekzadeh, M. Alizadeh, Parameter estimation of photovoltaic cells using improved Lozi map based chaotic optimization algorithm, Sol. Energy, 180 (2019), 180–191. https://doi.org/10.1016/j.solener.2019.01.026 doi: 10.1016/j.solener.2019.01.026
|
| [54] |
W. Long, T. Wu, M. Xu, M. Tang, S. Cai, Parameters identification of photovoltaic models by using an enhanced adaptive butterfly optimization algorithm, Energy, 229 (2021), 120750. https://doi.org/10.1016/j.energy.2021.120750 doi: 10.1016/j.energy.2021.120750
|
| [55] |
Y. Liu, A. A. Heidari, X. Ye, C. Chi, X. Zhao, C. Ma, et al., Evolutionary shuffled frog leaping with memory pool for parameter optimization, Energy Rep., 7 (2021), 584–606. https://doi.org/10.1016/j.egyr.2021.01.001 doi: 10.1016/j.egyr.2021.01.001
|
| [56] |
X. Chen, K. Yu, W. Du, W. Zhao, G. Liu, Parameters identification of solar cell models using generalized oppositional teaching learning based optimization, Energy, 99 (2016), 170–180. https://doi.org/10.1016/j.energy.2016.01.052 doi: 10.1016/j.energy.2016.01.052
|
| [57] |
K. Yu, X. Chen, X. Wang, Z. Wang, Parameters identification of photovoltaic models using self-adaptive teaching-learning-based optimization, Energy Convers. Manage., 145 (2017), 233–246. https://doi.org/10.1016/j.enconman.2017.04.054 doi: 10.1016/j.enconman.2017.04.054
|
| [58] |
K. Yu, J. J. Liang, B. Y. Qu, X. Chen, H. Wang, Parameters identification of photovoltaic models using an improved JAYA optimization algorithm, Energy Convers. Manage., 150 (2017), 742–753. https://doi.org/10.1016/j.enconman.2017.08.063 doi: 10.1016/j.enconman.2017.08.063
|
| [59] |
X. Chen, B. Xu, C. Mei, Y. Ding, K. Li, Teaching–learning–based artificial bee colony for solar photovoltaic parameter estimation, Appl. Energy, 212 (2018), 1578–1588. https://doi.org/10.1016/j.apenergy.2017.12.115 doi: 10.1016/j.apenergy.2017.12.115
|
| [60] |
S. M. Ebrahimi, E. Salahshour, M. Malekzadeh, F. Gordillo, Parameters identification of PV solar cells and modules using flexible particle swarm optimization algorithm, Energy, 179 (2019), 358–372. https://doi.org/10.1016/j.energy.2019.04.218 doi: 10.1016/j.energy.2019.04.218
|
| [61] |
Y. Zhang, C. Huang, Z. Jin, Backtracking search algorithm with reusing differential vectors for parameter identification of photovoltaic models, Energy Convers. Manage., 223 (2020), 113266. https://doi.org/10.1016/j.enconman.2020.113266 doi: 10.1016/j.enconman.2020.113266
|
| [62] |
Y. Zhang, M. Ma, Z. Jin, Comprehensive learning Jaya algorithm for parameter extraction of photovoltaic models, Energy, 211 (2020), 118644. https://doi.org/10.1016/j.energy.2020.118644 doi: 10.1016/j.energy.2020.118644
|
| [63] |
Y. Zhang, M. Ma, Z. Jin, Backtracking search algorithm with competitive learning for identification of unknown parameters of photovoltaic systems, Expert Syst. Appl., 160 (2020), 113750. https://doi.org/10.1016/j.eswa.2020.113750 doi: 10.1016/j.eswa.2020.113750
|
| [64] |
D. Oliva, M. Abd El Aziz, A. Ella Hassanien, Parameter estimation of photovoltaic cells using an improved chaotic whale optimization algorithm, Appl. Energy, 200 (2017), 141–154. https://doi.org/10.1016/j.apenergy.2017.05.029 doi: 10.1016/j.apenergy.2017.05.029
|
| [65] |
P. Lin, S. Cheng, W. Yeh, Z. Chen, L. Wu, Parameters extraction of solar cell models using a modified simplified swarm optimization algorithm, Sol. Energy, 144 (2017), 594–603. https://doi.org/10.1016/j.solener.2017.01.064 doi: 10.1016/j.solener.2017.01.064
|
| [66] |
G. Xiong, J. Zhang, D. Shi, Y. He, Parameter extraction of solar photovoltaic models using an improved whale optimization algorithm, Energy Convers. Manage., 174 (2018), 388–405. https://doi.org/10.1016/j.enconman.2018.08.053 doi: 10.1016/j.enconman.2018.08.053
|
| [67] |
A. M. Beigi, A. Maroosi, Parameter identification for solar cells and module using a Hybrid Firefly and Pattern Search Algorithms, Sol. Energy, 171 (2018), 435–446. https://doi.org/10.1016/j.solener.2018.06.092 doi: 10.1016/j.solener.2018.06.092
|
| [68] |
J. Liang, S. Ge, B. Qu, K. Yu, F. Liu, H. Yang, et al., Classified perturbation mutation based particle swarm optimization algorithm for parameters extraction of photovoltaic models, Energy Convers. Manage., 203 (2020), 112138. https://doi.org/10.1016/j.enconman.2019.112138 doi: 10.1016/j.enconman.2019.112138
|
| [69] |
X. Lin, Y. Wu, Parameters identification of photovoltaic models using niche-based particle swarm optimization in parallel computing architecture, Energy, 196 (2020), 117054. https://doi.org/10.1016/j.energy.2020.117054 doi: 10.1016/j.energy.2020.117054
|


Figures(10) / Tables(22)

Juan Zhao, Yujun Zhang, Shuijia Li, Yufei Wang, Yuxin Yan, Zhengming Gao. A chaotic self-adaptive JAYA algorithm for parameter extraction of photovoltaic models[J]. Mathematical Biosciences and Engineering, 2022, 19(6): 5638-5670. doi: 10.3934/mbe.2022264







 DownLoad:
DownLoad: