Citation: Qiaojun Situ, Jinzhi Lei. A mathematical model of stem cell regeneration with epigenetic state transitions[J]. Mathematical Biosciences and Engineering, 2017, 14(5&6): 1379-1397. doi: 10.3934/mbe.2017071

| [1] | [ R. C. Adam,H. Yang,S. Rockowitz,S. B. Larsen,M. Nikolova,D. S. Oristian,L. Polak,M. Kadaja,A. Asare,D. Zheng,E. Fuchs, Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice, Nature, 521 (2015): 366-370. |
| [2] | [ M. Adimy,F. Crauste,S. Ruan, A mathematical study of the hematopoiesis process with applications to chronic myelogenous leukemia, SIAM journal on applied mathematics, 65 (2005): 1328-1352. |
| [3] | [ M. Adimy,F. Crauste,S. Ruan, Stability and Hopf bifurcation in a mathematical model of pluripotent stem cell dynamics, Nonlinear Analysis: Real World Applications, 6 (2005): 651-670. |
| [4] | [ M. Adimy,F. Crauste,S. Ruan, Periodic oscillations in leukopoiesis models with two delays, J Theor Biol, 242 (2006): 288-299. |
| [5] | [ S. Bernard,J. Bélair,M. C. Mackey, Oscillations in cyclical neutropenia: New evidence based on mathematical modeling, J Theor Biol, 223 (2003): 283-298. |
| [6] | [ F. J. Burns,I. F. Tannock, On the existence of a G0-phase in the cell cycle, Cell Proliferation, 3 (1970): 321-334. |
| [7] | [ H. H. Chang,M. Hemberg,M. Barahona,D. E. Ingber,S. Huang, Transcriptome-wide noise controls lineage choice in mammalian progenitor cells, Nature, 453 (2008): 544-547. |
| [8] | [ S. J. Corey, M. Kimmel and J. N. Leonard (eds.), A Systems Biology Approach to Blood, vol. 844 of Advances in Experimental Medicine and Biology, Springer, London, 2014. |
| [9] | [ D. C. Dale,M. C. Mackey, Understanding, treating and avoiding hematological disease: better medicine through mathematics?, Bull Math Biol, 77 (2015): 739-757. |
| [10] | [ D. Dingli,A. Traulsen,J. M. Pacheco, Stochastic dynamics of hematopoietic tumor stem cells, Cell Cycle (Georgetown, Tex), 6 (2007): 461-466. |
| [11] | [ B. Dykstra,D. Kent,M. Bowie,L. McCaffrey,M. Hamilton,K. Lyons,S.-J. Lee,R. Brinkman,C. Eaves, Long-term propagation of distinct hematopoietic differentiation programs in vivo, Stem Cell, 1 (2007): 218-229. |
| [12] | [ T. M. Gibson,C. A. Gersbach, Single-molecule analysis of myocyte differentiation reveals bimodal lineage commitment, Integr Biol (Camb), 7 (2015): 663-671. |
| [13] | [ P. B. Gupta,C. M. Fillmore,G. Jiang,S. D. Shapira,K. Tao,C. Kuperwasser,E. S. Lander, Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells, Cell, 146 (2010): 633-644. |
| [14] | [ K. Hayashi,S. M. C. de Sousa Lopes,F. Tang,M. A. Surani, Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states, Stem Cell, 3 (2008): 391-401. |
| [15] | [ G. M. Hu,C. Y. Lee,Y.-Y. Chen,N. N. Pang,W. J. Tzeng, Mathematical model of heterogeneous cancer growth with an autocrine signalling pathway, Cell Prolif, 45 (2012): 445-455. |
| [16] | [ D. Huh,J. Paulsson, Non-genetic heterogeneity from stochastic partitioning at cell division, Nat Genet, 43 (2011): 95-100. |
| [17] | [ A. D. Lander,K. K. Gokoffski,F. Y. M. Wan,Q. Nie,A. L. Calof, Cell lineages and the logic of proliferative control, PLoS biology, 7 (2009): e15-e15. |
| [18] | [ J. Lei,S. A. Levin,Q. Nie, Mathematical model of adult stem cell regeneration with cross-talk between genetic and epigenetic regulation, Proc Natl Acad Sci USA, 111 (2014): E880-E887. |
| [19] | [ J. Lei,M. C. Mackey, Stochastic differential delay equation, moment stability, and application to hematopoietic stem cell regulation system, SIAM journal on applied mathematics, 67 (2007): 387-407. |
| [20] | [ J. Lei,M. C. Mackey, Multistability in an age-structured model of hematopoiesis: Cyclical neutropenia, J Theor Biol, 270 (2011): 143-153. |
| [21] | [ J. Lei,C. Wang, On the reducibility of compartmental matrices, Comput Biol Med, 38 (2008): 881-885. |
| [22] | [ B. D. MacArthur, Collective dynamics of stem cell populations, Proc Natl Acad Sci USA, 111 (2014): 3653-3654. |
| [23] | [ B. D. MacArthur,I. R. Lemischka, Statistical mechanics of pluripotency, Cell, 154 (2013): 484-489. |
| [24] | [ M. C. Mackey, Unified hypothesis for the origin of aplastic anemia and periodic hematopoiesis, Blood, 51 (1978): 941-956. |
| [25] | [ M. C. Mackey, Cell kinetic status of haematopoietic stem cells, Cell Prolif, 34 (2001): 71-83. |
| [26] | [ M. Mangel and M. B. Bonsall, Phenotypic evolutionary models in stem cell biology: Replacement, quiescence, and variability, PLoS ONE, 3 (2008), e1591. |
| [27] | [ M. Mangel,M. B. Bonsall, Stem cell biology is population biology: Differentiation of hematopoietic multipotent progenitors to common lymphoid and myeloid progenitors, Theor Biol Med Model, 10 (2012): 5-5. |
| [28] | [ C. S. Potten,M. Loeffler, Stem cells: Attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt, Development, 110 (1990): 1001-1020. |
| [29] | [ A. V. Probst,E. Dunleavy,G. Almouzni, Epigenetic inheritance during the cell cycle, Nat Rev Mol Cell Biol, 10 (2009): 192-206. |
| [30] | [ J. E. Purvis,K. W. Karhohs,C. Mock,E. Batchelor,A. Loewer,G. Lahav, p53 dynamics control cell fate, Science, 336 (2012): 1440-1444. |
| [31] | [ J. E. Purvis,G. Lahav, Encoding and decoding cellular information through signaling dynamics, Cell, 152 (2013): 945-956. |
| [32] | [ A. Rezza,Z. Wang,R. Sennett,W. Qiao,D. Wang,N. Heitman,K. W. Mok,C. Clavel,R. Yi,P. Zandstra,A. Ma'ayan,M. Rendl, Signaling networks among stem cell precursors, transit-amplifying progenitors, and their niche in developing hair follicles, Cell Rep, 14 (2016): 3001-3018. |
| [33] | [ I. Rodriguez-Brenes,N. Komarova,D. Wodarz, Evolutionary dynamics of feedback escape and the development of stem-cell–driven cancers, Proc Natl Acad Sci USA, 108 (2011): 18983-18988. |
| [34] | [ P. Rué,A. Martinez-Arias, Cell dynamics and gene expression control in tissue homeostasis and development, Mol Syst Biol, 11 (2015): 792-792. |
| [35] | [ T. Schepeler,M. E. Page,K. B. Jensen, Heterogeneity and plasticity of epidermal stem cells, Development, 141 (2014): 2559-2567. |
| [36] | [ Z. S. Singer,J. Yong,J. Tischler,J. A. Hackett,A. Altinok,M. A. Surani,L. Cai,M. B. Elowitz, Dynamic heterogeneity and DNA methylation in embryonic stem cells, Mol Cell, 55 (2014): 319-331. |
| [37] | [ K. Takaoka and H. Hamada, Origin of cellular asymmetries in the pre-implantation mouse embryo: A hypothesis, Philos Trans R Soc Lond B Biol Sci, 369 (2014). |
| [38] | [ J. E. Till, E. A. McCulloch and L. Siminovitch, A Stochastic Model of Stem Cell Proliferation, Based on the Growth of Spleen Colony-Forming Cells, in Proceedings of the National Academy of Sciences of the United States of America, 1963, 29–36. |
| [39] | [ A. Traulsen,T. Lenaerts,J. M. Pacheco,D. Dingli, On the dynamics of neutral mutations in a mathematical model for a homogeneous stem cell population, Journal of the Royal Society, Interface / the Royal Society, 10 (2013): 20120810-20120810. |
| [40] | [ H. Wu,Y. Zhang, Reversing DNA methylation: Mechanisms, genomics, and biological functions, Cell, 156 (2014): 45-68. |
| [41] | [ M. Zernicka-Goetz,S. A. Morris,A. W. Bruce, Making a firm decision: Multifaceted regulation of cell fate in the early mouse embryo, Nat Rev Genet, 10 (2009): 467-477. |
| [42] | [ X.-P. Zhang,F. Liu,Z. Cheng,W. Wang, Cell fate decision mediated by p53 pulses, Proc Natl Acad Sci USA, 106 (2009): 12245-12250. |
| [43] | [ D. Zhou,D. Wu,Z. Li,M. Qian,M. Q. Zhang, Population dynamics of cancer cells with cell state conversions, Quant Biol, 1 (2013): 201-208. |
| [44] | [ C. Zhuge,X. Sun,J. Lei, On positive solutions and the Omega limit set for a class of delay differential equations, DCDS-B, 18 (2013): 2487-2503. |
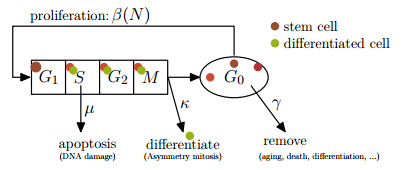
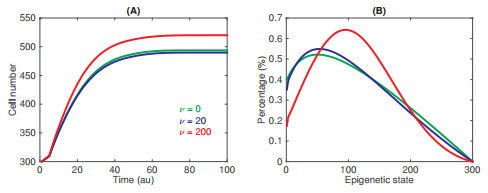
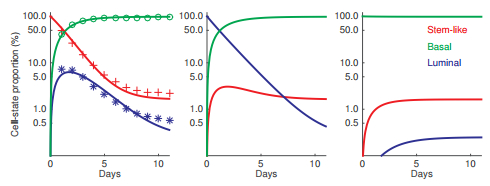
Figures(3) / Tables(1)

Qiaojun Situ, Jinzhi Lei. A mathematical model of stem cell regeneration with epigenetic state transitions[J]. Mathematical Biosciences and Engineering, 2017, 14(5&6): 1379-1397. doi: 10.3934/mbe.2017071







 DownLoad:
DownLoad: