Citation: Arnab Chanda, Rebecca Graeter, Vinu Unnikrishnan. Effect of blasts on subject-specific computational models of skin and bone sections at various locations on the human body[J]. AIMS Materials Science, 2015, 2(4): 425-447. doi: 10.3934/matersci.2015.4.425

| [1] |
Kulkarni S, Gao X-L, Horner S, et al. (2013) Ballistic helmets-Their design, materials, and performance against traumatic brain injury. Compos Struct 101: 313-331. doi: 10.1016/j.compstruct.2013.02.014

|
| [2] |
Jenson D, Unnikrishnan VU (2015) Energy dissipation of nanocomposite based helmets for blast-induced traumatic brain injury mitigation. Compos Struct 121: 211-216. doi: 10.1016/j.compstruct.2014.08.028
|
| [3] | Stuhmiller JH, Phillips Y, Richmond D (1991) The physics and mechanisms of primary blast injury. Conventional warfare: ballistic, blast, and burn injuries Washington, DC: Office of the Surgeon General of the US Army, 241-270. |
| [4] | Born CT (2004) Blast trauma: the fourth weapon of mass destruction. Scandinavian journal of surgery: SJS: official organ for the Finnish Surgical Society and the Scandinavian Surgical Society 94: 279-285. |
| [5] |
Chandra N, Ganpule S, Kleinschmit N, et al. (2012) Evolution of blast wave profiles in simulated air blasts: experiment and computational modeling. Shock Waves 22: 403-415. doi: 10.1007/s00193-012-0399-2
|
| [6] | Chandra N, Skotak M, Wang F, et al. (2013) Do Primary Blast-Shock Waves Cause Mild TBI? Experimental Evidence Based On Animal Models And Human Cadaveric Heads. Mary Ann Liebert, Inc 140 Huguenot Street, 3rd Fl, New Rochelle, NY 10801 USA, A80-A80. |
| [7] | Ganpule SG, Chandra N, Salzar R (2013) Mechanics Of Blast Loading On Post-Mortem Human Heads in The Study Of Traumatic Brain Injury (TBI) Using Experimental And Computational Approaches [PhD Dissertation]: University of Nebraska-Lincoln, 289. |
| [8] | Ganpule S, Chandra N (2013) Mechanics of Interaction of Blast Waves on Surrogate Head: Effect of Head Orientation. ASME 2013 Summer Bioengineering Conference: American Society of Mechanical Engineers, V01BT55A028-029. |
| [9] | Kangarlou K (2013) Mechanics of Blast Loading on the Head Models in the Study of Traumatic Brain Injury. Nationalpark-Forschung In Der Schweiz (Switzerland Research Park Journal), 102. |
| [10] | Sundaramurthy A, Alai A, Ganpule S, et al. (2012) Blast-induced biomechanical loading of the rat: an experimental and anatomically accurate computational blast injury model. J Neurotraum 29: 2352-2364. |
| [11] |
Chafi MS, Ganpule S, Gu L, et al. (2011) Dynamic response of brain subjected to blast loadings: influence of frequency ranges. Int J Appl Mech 3: 803-823. doi: 10.1142/S175882511100124X
|
| [12] | Ganpule S, Gu L, Alai A, et al. (2012) Role of helmet in the mechanics of shock wave propagation under blast loading conditions. Computer Methods Biomec 15: 1233-1244. |
| [13] | Ganpule S, Gu L, Cao G, et al. (2009) The effect of shock wave on a human head. ASME 2009 International Mechanical Engineering Congress and Exposition: American Society of Mechanical Engineers, 339-346. |
| [14] | Ganpule S, Gu L, Chandra N (2010) MRI-based three dimensional modeling of blast traumatic brain injury (bTBI). ASME 2010 International Mechanical Engineering Congress and Exposition: American Society of Mechanical Engineers, 181-183. |
| [15] |
Gu L, Chafi MS, Ganpule S, et al. (2012) The influence of heterogeneous meninges on the brain mechanics under primary blast loading. Compos Part B-Eng 43: 3160-3166. doi: 10.1016/j.compositesb.2012.04.014
|
| [16] | Gupta RK, Przekwas A (2013) Mathematical models of blast-induced TBI: current status, challenges, and prospects. Front Neurol 4: 59. |
| [17] |
Hayda R, Harris RM, Bass CD (2004) Blast injury research: modeling injury effects of landmines, bullets, and bombs. Clin Orthop Relate R 422: 97-108. doi: 10.1097/01.blo.0000128295.28666.ee
|
| [18] | Jenson D, Unnikrishnan V (2014) Multiscale Simulation of Ballistic Composites for Blast Induced Traumatic Brain Injury Mitigation. ASME 2014 International Mechanical Engineering Congress and Exposition: American Society of Mechanical Engineers. V009T012A072-V009T012A077. |
| [19] | Hull J (1992) Traumatic amputation by explosive blast: pattern of injury in survivors. Brit J Surg 79: 1303-1306. |
| [20] |
DePalma RG, Burris DG, Champion HR, et al. (2005) Blast injuries. New Engl J Med 352: 1335-1342. doi: 10.1056/NEJMra042083
|
| [21] |
Wightman JM, Gladish SL (2001) Explosions and blast injuries. Ann Emerg Med 37: 664-678. doi: 10.1067/mem.2001.114906
|
| [22] |
Gondusky JS, Reiter MP (2005) Protecting military convoys in Iraq: an examination of battle injuries sustained by a mechanized battalion during Operation Iraqi Freedom II. Mil Med 170: 546. doi: 10.7205/MILMED.170.6.546
|
| [23] | Beekley AC, Blackbourne LH, Sebesta JA, et al. (2008) Selective nonoperative management of penetrating torso injury from combat fragmentation wounds. J Trauma Acute Care 64: S108-S117. |
| [24] |
Xydakis MS, Fravell MD, Nasser KE, et al. (2005) Analysis of battlefield head and neck injuries in Iraq and Afghanistan. Otolaryng Head Neck 133: 497-504. doi: 10.1016/j.otohns.2005.07.003
|
| [25] |
Breeze J, Allanson-Bailey LS, Hunt NC, et al. (2012) Mortality and morbidity from combat neck injury. J Trauma Acute Care 72: 969-974. doi: 10.1097/TA.0b013e31823e20a0
|
| [26] | Dussault MC (2013) Blast injury to the human skeleton: recognition, identification and differentiation using morphological and statistical approaches [PhD dissertation]: Bournemouth University, School of Applied Sciences, 350. |
| [27] | Bertucci R, Liao J, Williams L (2011) Development of a Lower Extremity Model for Finite Element Analysis at Blast Condition. ASME 2011 Summer Bioengineering Conference: American Society of Mechanical Engineers, 1035-1036. |
| [28] | Netter FH (2014) Atlas of human anatomy. Philadelphia, PA: Elsevier Health Sciences. |
| [29] | Wolpoff MH, Caspari R (1997) Race and human evolution. New York, NY: Simon and Schuster. |
| [30] |
Barker DE (1951) Skin thickness in the human. Plast Reconstr Surg 7: 115-116. doi: 10.1097/00006534-195102000-00004
|
| [31] |
Domaracki M, Stephan CN (2006) Facial Soft Tissue Thicknesses in Australian Adult Cadavers*. J Forensic Sci 51: 5-10. doi: 10.1111/j.1556-4029.2005.00009.x
|
| [32] | Fields ML, Greenberg BH, Burkett LL (1967) Roentgenographic measurement of skin and heel-pad thickness in the diagnosis of acromegaly. Am J Med Sci 254: 528-533. |
| [33] | Garn SM, Haskell JA (1960) Fat thickness and developmental status in childhood and adolescence. AM J Dis Child 99: 746-751. |
| [34] |
Holbrook KA, Odland GF (1974) Regional differences in the thickness (cell layers) of the human stratum corneum: an ultrastructural analysis. J Invest Dermatol 62: 415-422. doi: 10.1111/1523-1747.ep12701670
|
| [35] |
Hwang HS, Park MK, Lee WJ, et al. (2012) Facial soft tissue thickness database for craniofacial reconstruction in Korean adults. J Forensic Sci 57: 1442-1447. doi: 10.1111/j.1556-4029.2012.02192.x
|
| [36] |
Lee Y, Hwang K (2002) Skin thickness of Korean adults. Surg Radiol Anat 24: 183-189. doi: 10.1007/s00276-002-0034-5
|
| [37] |
Sahni D, Singh G, Jit I, et al. (2008) Facial soft tissue thickness in northwest Indian adults. Forensic Sci Int 176: 137-146. doi: 10.1016/j.forsciint.2007.07.012
|
| [38] |
Simpson E, Henneberg M (2002) Variation in soft tissue thicknesses on the human face and their relation to craniometric dimensions. Am J Phys Anthropol 118: 121-133. doi: 10.1002/ajpa.10073
|
| [39] | Sipahioğlu S, Ulubay H, Diren HB (2012) Midline facial soft tissue thickness database of Turkish population: MRI study. Forensic Sci Int 219, 1-282: e1-e8. |
| [40] |
Southwood W (1955) The thickness of the skin. Plast Reconstr Surg 15: 423-429. doi: 10.1097/00006534-195505000-00006
|
| [41] | Tedeschi-Oliveira SV, Melani RFH, de Almeida NH, et al. (2009) Facial soft tissue thickness of Brazilian adults. Forensic Sci Int 193, 1-127: e1-e7. |
| [42] |
Whitmore SE, Sago NJG (2000) Caliper-measured skin thickness is similar in white and black women. J Am Acad Dermatol 42: 76-79. doi: 10.1016/S0190-9622(00)90012-4
|
| [43] |
Whitton JT (1973) New values for epidermal thickness and their importance. Health Phys 24: 1-8. doi: 10.1097/00004032-197301000-00001
|
| [44] |
Chanda A, Unnikrishnan V, Roy S, et al. (2015) Computational Modeling of the Female Pelvic Support Structures and Organs to Understand the Mechanism of Pelvic Organ Prolapse: A Review. Appl Mech Rev 67: 040801. doi: 10.1115/1.4030967
|
| [45] |
Chanda A, Ghoneim H (2015) Pumping potential of a two-layer left-ventricle-like flexible-matrix-composite structure. Compos Struct 122: 570-575. doi: 10.1016/j.compstruct.2014.11.069
|
| [46] | Gray H (1918) Anatomy of the human body. Baltimore, MD: Lea & Febiger. |
| [47] |
Martins P, Natal Jorge R, Ferreira A (2006) A Comparative Study of Several Material Models for Prediction of Hyperelastic Properties: Application to Silicone Rubber and Soft Tissues. Strain 42: 135-147. doi: 10.1111/j.1475-1305.2006.00257.x
|
| [48] |
Annaidh AN, Bruyère K, Destrade M, et al. (2012) Characterization of the anisotropic mechanical properties of excised human skin. J Mech Behav Biomed 5: 139-148. doi: 10.1016/j.jmbbm.2011.08.016
|
| [49] | Chanda A, Unnikrishnan V, Flynn Z (2015) Biofidelic Human Skin Simulant. US Patent Application No 62/189, 504. |
| [50] |
Kaster T, Sack I, Samani A (2011) Measurement of the hyperelastic properties of ex vivo brain tissue slices. J Biomech 44: 1158-1163. doi: 10.1016/j.jbiomech.2011.01.019
|
| [51] |
O'Hagan JJ, Samani A (2009) Measurement of the hyperelastic properties of 44 pathological ex vivo breast tissue samples. Phys Med Biol 54: 2557. doi: 10.1088/0031-9155/54/8/020
|
| [52] | Flynn CO (2007) The design and validation of a multi-layer model of human skin [PhD Dissertation]. Sligo, Ireland: Institute of Technology, Sligo. |
| [53] | Adegbenro A, Taylor S (2013) Structural, Physiological, Functional, and Cultural Differences in Skin of Color. Skin of Color: Springer, 1-19. |
| [54] | Saggar S, Wesley NO, Moulton-Levy NM, et al. (2007) Ethnic differences in skin properties: the objective data. Boca Raton, FL: Taylor & Francis. |
| [55] |
Wesley NO, Maibach HI (2003) Racial (ethnic) differences in skin properties. Am J Clin Dermatol 4: 843-860. doi: 10.2165/00128071-200304120-00004
|
| [56] | Yosipovitch G, Theng C (2002) Asian skin: its architecture, function and differences from Caucasian skin. Cosmetics Toiletries 117: 57-62. |

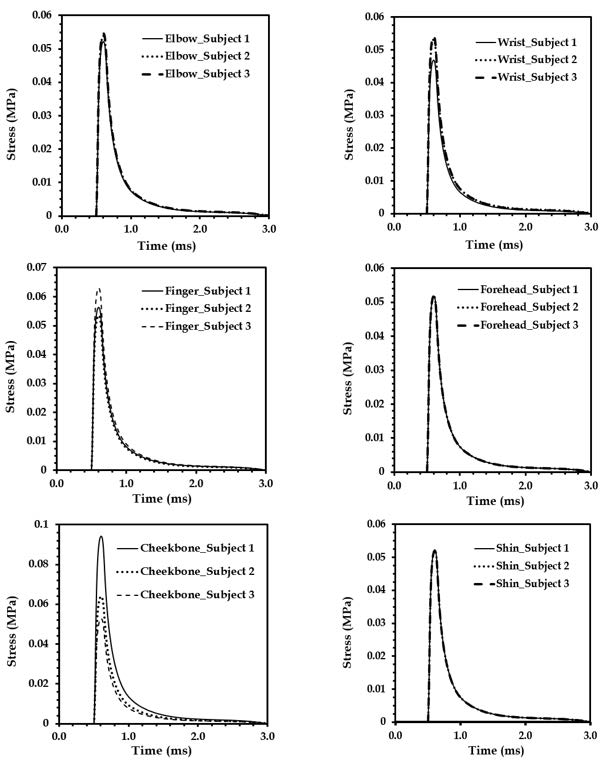
Figures(18) / Tables(4)

Arnab Chanda, Rebecca Graeter, Vinu Unnikrishnan. Effect of blasts on subject-specific computational models of skin and bone sections at various locations on the human body[J]. AIMS Materials Science, 2015, 2(4): 425-447. doi: 10.3934/matersci.2015.4.425







 DownLoad:
DownLoad: