Citation: Stephanie H. DeLuca, Samuel L. DeLuca, Andrew Leaver-Fay, Jens Meiler. RosettaTMH: a method for membrane protein structure elucidation combining EPR distance restraints with assembly of transmembrane helices[J]. AIMS Biophysics, 2016, 3(1): 1-26. doi: 10.3934/biophy.2016.1.1

| [1] |
Krishnamurthy H, Gouaux E (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481: 469–474. doi: 10.1038/nature10737

|
| [2] | Sanders CR, Sonnichsen F (2006) Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem 44 Spec No: S24–40. |
| [3] |
Horst R, Stanczak P, Stevens RC, et al. (2013) beta2-Adrenergic receptor solutions for structural biology analyzed with microscale NMR diffusion measurements. Angew Chem Int Ed Engl 52: 331–335. doi: 10.1002/anie.201205474
|
| [4] |
Chun E, Thompson AA, Liu W, et al. (2012) Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20: 967–976. doi: 10.1016/j.str.2012.04.010
|
| [5] |
Baker LA, Baldus M (2014) Characterization of membrane protein function by solid-state NMR spectroscopy. Curr Opin Struct Biol 27: 48–55. doi: 10.1016/j.sbi.2014.03.009
|
| [6] |
Hopkins AL, Groom CR (2002) The druggable genome. Nat Rev Drug Discov 1: 727–730. doi: 10.1038/nrd892
|
| [7] |
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5: 993–996. doi: 10.1038/nrd2199
|
| [8] |
Bakheet TM, Doig AJ (2009) Properties and identification of human protein drug targets. Bioinformatics 25: 451–457. doi: 10.1093/bioinformatics/btp002
|
| [9] |
Maslennikov I, Choe S (2013) Advances in NMR structures of integral membrane proteins. Curr Opin Struct Biol 23: 555–562. doi: 10.1016/j.sbi.2013.05.002
|
| [10] |
Klammt C, Maslennikov I, Bayrhuber M, et al. (2012) Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat Methods 9: 834–839. doi: 10.1038/nmeth.2033
|
| [11] |
Berman HM, Battistuz T, Bhat TN, et al. (2002) The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 58: 899–907. doi: 10.1107/S0907444902003451
|
| [12] |
Berman HM, Westbrook J, Feng Z, et al. (2000) The Protein Data Bank. Nucleic Acids Res 28: 235–242. doi: 10.1093/nar/28.1.235
|
| [13] |
Tang M, Comellas G, Rienstra CM (2013) Advanced solid-state NMR approaches for structure determination of membrane proteins and amyloid fibrils. Acc Chem Res 46: 2080–2088. doi: 10.1021/ar4000168
|
| [14] |
Ni QZ, Daviso E, Can TV, et al. (2013) High frequency dynamic nuclear polarization. Acc Chem Res 46: 1933–1941. doi: 10.1021/ar300348n
|
| [15] |
Zou P, McHaourab HS (2010) Increased sensitivity and extended range of distance measurements in spin-labeled membrane proteins: Q-band double electron-electron resonance and nanoscale bilayers. Biophys J 98: L18–20. doi: 10.1016/j.bpj.2009.12.4193
|
| [16] |
Mchaourab HS, Steed PR, Kazmier K (2011) Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure 19: 1549–1561. doi: 10.1016/j.str.2011.10.009
|
| [17] |
Tusnady GE, Dosztanyi Z, Simon I (2004) Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics 20: 2964–2972. doi: 10.1093/bioinformatics/bth340
|
| [18] | Mchaourab HS, Lietzow MA, Hideg K, et al. (1996) Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 35: 7692–7704. |
| [19] |
Hubbell WL, McHaourab HS, Altenbach C, et al. (1996) Watching proteins move using site-directed spin labeling. Structure 4: 779–783. doi: 10.1016/S0969-2126(96)00085-8
|
| [20] |
Fanucci GE, Cafiso DS (2006) Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol 16: 644–653. doi: 10.1016/j.sbi.2006.08.008
|
| [21] | Weierstall U, James D, Wang C, et al. (2014) Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun 5: 3309. |
| [22] |
Liu W, Wacker D, Gati C, et al. (2013) Serial femtosecond crystallography of G protein-coupled receptors. Science 342: 1521–1524. doi: 10.1126/science.1244142
|
| [23] | Li D, Boland C, Walsh K, et al. (2012) Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. J Vis Exp: e4000. |
| [24] | Li D, Boland C, Aragao D, et al. (2012) Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. J Vis Exp: e4001. |
| [25] |
Alexander N, Al-Mestarihi A, Bortolus M, et al. (2008) De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure 16: 181–195. doi: 10.1016/j.str.2007.11.015
|
| [26] |
Hirst SJ, Alexander N, Mchaourab HS, et al. (2011) RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J Struct Biol 173: 506–514. doi: 10.1016/j.jsb.2010.10.013
|
| [27] |
Islam SM, Stein RA, McHaourab HS, et al. (2013) Structural refinement from restrained-ensemble simulations based on EPR/DEER data: application to T4 lysozyme. J Phys Chem B 117: 4740–4754. doi: 10.1021/jp311723a
|
| [28] |
Fischer AW, Alexander NS, Woetzel N, et al. (2015) BCL::MP-Fold: Membrane protein structure prediction guided by EPR restraints. Proteins 83: 1947–1962. doi: 10.1002/prot.24801
|
| [29] |
Jeschke G, Chechik V, Ionita P, et al. (2006) DeerAnalysis2006 - a comprehensive software package for analyzing pulsed ELDOR data. Appl Magn Reson 30: 473–498. doi: 10.1007/BF03166213
|
| [30] |
Hagelueken G, Ward R, Naismith JH, et al. (2012) MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl Magn Reson 42: 377–391. doi: 10.1007/s00723-012-0314-0
|
| [31] |
Beasley KN, Sutch BT, Hatmal MM, et al. (2015) Computer Modeling of Spin Labels: NASNOX, PRONOX, and ALLNOX. Methods Enzymol 563: 569–593. doi: 10.1016/bs.mie.2015.07.021
|
| [32] |
Hatmal MM, Li Y, Hegde BG, et al. (2012) Computer modeling of nitroxide spin labels on proteins. Biopolymers 97: 35–44. doi: 10.1002/bip.21699
|
| [33] |
Alexander NS, Stein RA, Koteiche HA, et al. (2013) RosettaEPR: Rotamer Library for Spin Label Structure and Dynamics. PLoS One 8: e72851. doi: 10.1371/journal.pone.0072851
|
| [34] |
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815. doi: 10.1006/jmbi.1993.1626
|
| [35] |
Fiser A, Sali A (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374: 461–491. doi: 10.1016/S0076-6879(03)74020-8
|
| [36] |
Webb B, Sali A (2014) Protein structure modeling with MODELLER. Methods Mol Biol 1137: 1–15. doi: 10.1007/978-1-4939-0366-5_1
|
| [37] | Webb B, Sali A (2014) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 47: 5.6.1–5.6.32. |
| [38] |
Rohl CA, Strauss CEM, Chivian D, et al. (2004) Modeling structurally variable regions in homologous proteins with rosetta. Proteins 55: 656–677. doi: 10.1002/prot.10629
|
| [39] |
Misura KM, Chivian D, Rohl CA, et al. (2006) Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc Natl Acad Sci U S A 103: 5361–5366. doi: 10.1073/pnas.0509355103
|
| [40] |
Schwede T, Kopp J, Guex N, et al. (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 31: 3381–3385. doi: 10.1093/nar/gkg520
|
| [41] | Zhang Y (2009) I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77 Suppl 9: 100–113. |
| [42] |
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. doi: 10.1186/1471-2105-9-40
|
| [43] |
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738. doi: 10.1038/nprot.2010.5
|
| [44] |
Combs SA, Deluca SL, Deluca SH, et al. (2013) Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc 8: 1277–1298. doi: 10.1038/nprot.2013.074
|
| [45] | Stevens RC, Cherezov V, Katritch V, et al. (2013) The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov 12: 25–34. |
| [46] |
Kroeze WK, Sheffler DJ, Roth BL (2003) G-protein-coupled receptors at a glance. J Cell Sci 116: 4867–4869. doi: 10.1242/jcs.00902
|
| [47] |
Yamashita A, Singh SK, Kawate T, et al. (2005) Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437: 215–223. doi: 10.1038/nature03978
|
| [48] |
Faham S, Watanabe A, Besserer GM, et al. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321: 810–814. doi: 10.1126/science.1160406
|
| [49] |
Perez C, Koshy C, Yildiz O, et al. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature 490: 126–130. doi: 10.1038/nature11403
|
| [50] |
Ma D, Lu P, Yan C, et al. (2012) Structure and mechanism of a glutamate-GABA antiporter. Nature 483: 632–636. doi: 10.1038/nature10917
|
| [51] |
Kazmier K, Sharma S, Quick M, et al. (2014) Conformational dynamics of ligand-dependent alternating access in LeuT. Nat Struct Mol Biol 21: 472–479. doi: 10.1038/nsmb.2816
|
| [52] |
Gregory KJ, Nguyen ED, Reiff SD, et al. (2013) Probing the metabotropic glutamate receptor 5 (mGlu(5)) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a "molecular switch" in PAM pharmacology. Mol Pharmacol 83: 991–1006. doi: 10.1124/mol.112.083949
|
| [53] | Yarov-Yarovoy V, Schonbrun J, Baker D (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62: 1010–1025. |
| [54] |
Barth P, Schonbrun J, Baker D (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A 104: 15682–15687. doi: 10.1073/pnas.0702515104
|
| [55] |
Barth P, Wallner B, Baker D (2009) Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A 106: 1409–1414. doi: 10.1073/pnas.0808323106
|
| [56] |
Nugent T, Jones DT (2012) Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc Natl Acad Sci U S A 109: E1540–1547. doi: 10.1073/pnas.1120036109
|
| [57] |
Nugent T, Jones DT (2013) Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinformatics 14: 276. doi: 10.1186/1471-2105-14-276
|
| [58] |
Hopf TA, Colwell LJ, Sheridan R, et al. (2012) Three-dimensional structures of membrane proteins from genomic sequencing. Cell 149: 1607–1621. doi: 10.1016/j.cell.2012.04.012
|
| [59] |
Weiner BE, Woetzel N, Karakas M, et al. (2013) BCL::MP-fold: folding membrane proteins through assembly of transmembrane helices. Structure 21: 1107–1117. doi: 10.1016/j.str.2013.04.022
|
| [60] |
Simons KT, Kooperberg C, Huang E, et al. (1997) Assembly of Protein Tertiary Structures from Fragments with Similar Local Sequences using Simulated Annealing and Bayesian Scoring Functions. J Mol Biol 268: 209–225. doi: 10.1006/jmbi.1997.0959
|
| [61] |
Rohl CA, Strauss CE, Misura KM, et al. (2004) Protein structure prediction using Rosetta. Methods Enzymol 383: 66–93. doi: 10.1016/S0076-6879(04)83004-0
|
| [62] |
Kazmier K, Alexander NS, Meiler J, et al. (2011) Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. J Struct Biol 173: 549–557. doi: 10.1016/j.jsb.2010.11.003
|
| [63] |
Dimaio F, Leaver-Fay A, Bradley P, et al. (2011) Modeling symmetric macromolecular structures in rosetta3. PLoS One 6: e20450. doi: 10.1371/journal.pone.0020450
|
| [64] |
Leaver-Fay A, Tyka M, Lewis SM, et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487: 545–574. doi: 10.1016/B978-0-12-381270-4.00019-6
|
| [65] |
Metropolis N, Rosenbluth A, Rosenbluth M, et al. (1953) Equations of state calculations by fast computing machines. J Chem Phys 21: 1087–1091. doi: 10.1063/1.1699114
|
| [66] |
Metropolis NU, Ulam S (1949) The Monte Carlo Method. J Am Stat Assoc 44: 335–341. doi: 10.1080/01621459.1949.10483310
|
| [67] |
Perozo E, Cortes DM, Cuello LG (1999) Structural rearrangements underlying K+-channel activation gating. Science 285: 73–78. doi: 10.1126/science.285.5424.73
|
| [68] |
Liu YS, Sompornpisut P, Perozo E (2001) Structure of the KcsA channel intracellular gate in the open state. Nat Struct Biol 8: 883–887. doi: 10.1038/nsb1001-883
|
| [69] |
Zou P, Mchaourab HS (2009) Alternating Access of the Putative Substrate-Binding Chamber in the ABC Transporter MsbA. J Mol Biol 393: 574–585. doi: 10.1016/j.jmb.2009.08.051
|
| [70] |
Altenbach C, Cai K, Klein-Seetharaman J, et al. (2001) Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306-319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry 40: 15483–15492. doi: 10.1021/bi011546g
|
| [71] |
Viklund H, Elofsson A (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24: 1662–1668. doi: 10.1093/bioinformatics/btn221
|
| [72] |
Adamian L, Liang J (2006) Prediction of transmembrane helix orientation in polytopic membrane proteins. BMC Struct Biol 6: 13. doi: 10.1186/1472-6807-6-13
|
| [73] | Okada T, Sugihara M, Bondar AN, et al. (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol 342: 571–583. |
| [74] |
Misura KM, Baker D (2005) Progress and challenges in high-resolution refinement of protein structure models. Proteins 59: 15–29. doi: 10.1002/prot.20376
|
| [75] |
Tyka MD, Jung K, Baker D (2012) Efficient sampling of protein conformational space using fast loop building and batch minimization on highly parallel computers. J Comput Chem 33: 2483–2491. doi: 10.1002/jcc.23069
|
| [76] |
Woetzel N, Karakas M, Staritzbichler R, et al. (2012) BCL::Score--knowledge based energy potentials for ranking protein models represented by idealized secondary structure elements. PLoS One 7: e49242. doi: 10.1371/journal.pone.0049242
|
| [77] |
Karakas M, Woetzel N, Staritzbichler R, et al. (2012) BCL::Fold--de novo prediction of complex and large protein topologies by assembly of secondary structure elements. PLoS One 7: e49240. doi: 10.1371/journal.pone.0049240
|
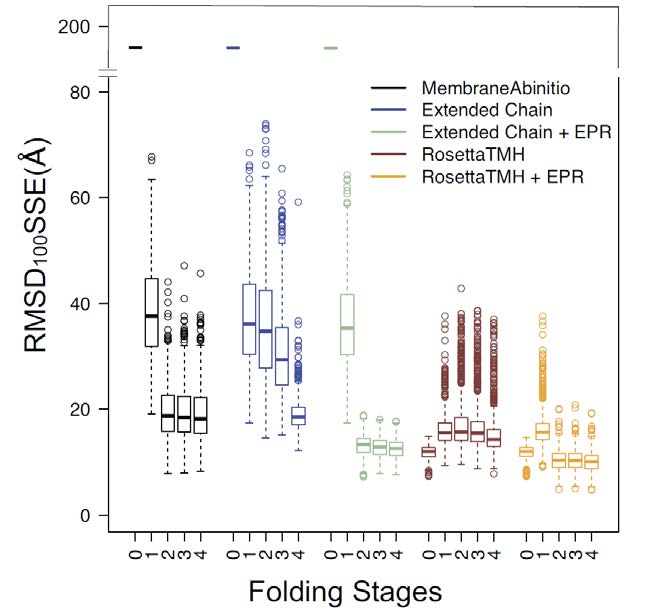
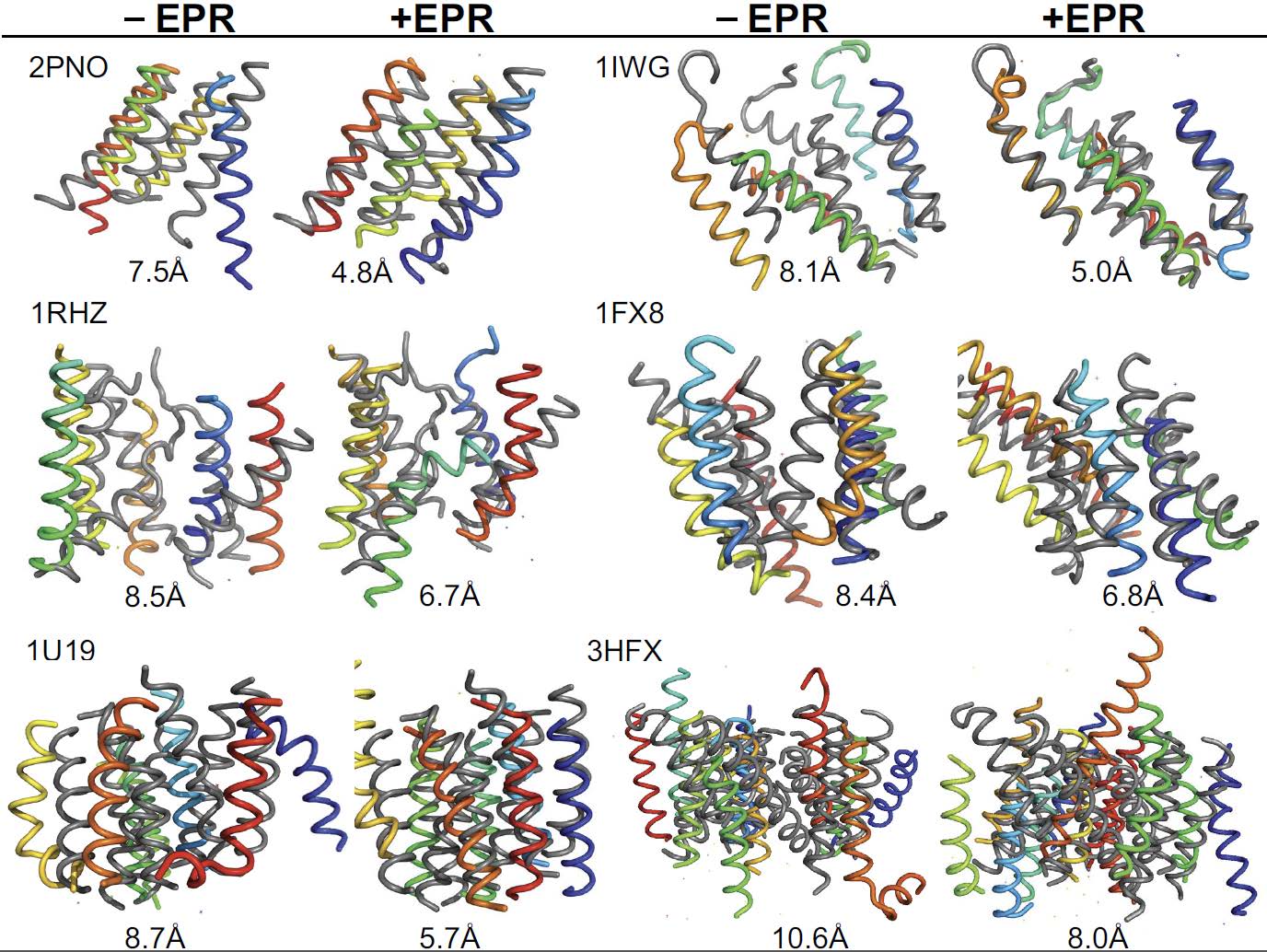
Figures(6) / Tables(2)

Stephanie H. DeLuca, Samuel L. DeLuca, Andrew Leaver-Fay, Jens Meiler. RosettaTMH: a method for membrane protein structure elucidation combining EPR distance restraints with assembly of transmembrane helices[J]. AIMS Biophysics, 2016, 3(1): 1-26. doi: 10.3934/biophy.2016.1.1







 DownLoad:
DownLoad: