Citation: Riyaz A. Mir, Jeff Lovelace, Nicholas P. Schafer, Peter D. Simone, Admir Kellezi, Carol Kolar, Gaelle Spagnol, Paul L. Sorgen, Hamid Band, Vimla Band, Gloria E. O. Borgstahl. Biophysical characterization and modeling of human Ecdysoneless (ECD) protein supports a scaffolding function[J]. AIMS Biophysics, 2016, 3(1): 195-210. doi: 10.3934/biophy.2016.1.195

| [1] |
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57-70. doi: 10.1016/S0092-8674(00)81683-9

|
| [2] |
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-74. doi: 10.1016/j.cell.2011.02.013
|
| [3] |
Frolov MV, Dyson NJ (2004) Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci 117: 2173-81. doi: 10.1242/jcs.01227
|
| [4] |
Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245-62. doi: 10.1101/gad.12.15.2245
|
| [5] |
Kim JH, Gurumurthy CB, Naramura M,et al. (2009) Role of mammalian Ecdysoneless in cell cycle regulation. J Biol Chem 284: 26402-10. doi: 10.1074/jbc.M109.030551
|
| [6] |
Henrich VC, Livingston L, Gilbert LI (1993) Developmental requirements for the ecdysoneless (ecd) locus in Drosophila melanogaster. Dev Genet 14: 369-77. doi: 10.1002/dvg.1020140506
|
| [7] |
Gaziova I, Bonnette PC, Henrich VC, et al. (2004) Cell-autonomous roles of the ecdysoneless gene in Drosophila development and oogenesis. Development 131: 2715-25. doi: 10.1242/dev.01143
|
| [8] |
Zhang Y, Chen J, Gurumurthy CB, et al. (2006) The human orthologue of Drosophila ecdysoneless protein interacts with p53 and regulates its function. Cancer Res 66: 7167-75. doi: 10.1158/0008-5472.CAN-06-0722
|
| [9] |
Zhao X, Mirza S, Alshareeda A, et al. (2012) Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: a marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast Cancer Res Treat 134: 171-80. doi: 10.1007/s10549-011-1946-8
|
| [10] |
Dey P, Rachagani S, Chakraborty S, et al. (2012) Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin Cancer Res 18: 6188-98. doi: 10.1158/1078-0432.CCR-12-1789
|
| [11] |
Bele A, Mirza S, Zhang Y, et al. (2015) The cell cycle regulator ecdysoneless cooperates with H-Ras to promote oncogenic transformation of human mammary epithelial cells. Cell Cycle 14: 990-1000. doi: 10.1080/15384101.2015.1006982
|
| [12] |
Horejsi Z, Stach L, Flower TG, et al. (2014) Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell Rep 7: 19-26. doi: 10.1016/j.celrep.2014.03.013
|
| [13] |
Kakihara Y, Houry WA (2012) The R2TP complex: discovery and functions. Biochim Biophys Acta 1823: 101-7. doi: 10.1016/j.bbamcr.2011.08.016
|
| [14] |
Boulon S, Bertrand E, Pradet-Balade B (2012) HSP90 and the R2TP co-chaperone complex: building multi-protein machineries essential for cell growth and gene expression. RNA Biol 9: 148-54. doi: 10.4161/rna.18494
|
| [15] |
Horejsi Z, Takai H, Adelman CA, et al. (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell 39: 839-50. doi: 10.1016/j.molcel.2010.08.037
|
| [16] | Mir RA, Bele A, Mirza S, et al. (2015) A novel interaction of ECD protein with R2TP complex component RUVBL1 is required for the functional role of ECD in cell cycle progression. Mol Cell Biol 36: 886-99. |
| [17] |
Suh HW, Yun S, Song H, et al. (2013) TXNIP interacts with hEcd to increase p53 stability and activity. Biochem Biophys Res Commun 438: 264-9. doi: 10.1016/j.bbrc.2013.07.036
|
| [18] |
Claudius AK, Romani P, Lamkemeyer T, et al. (2014) Unexpected role of the steroid-deficiency protein ecdysoneless in pre-mRNA splicing. PLoS Genet 10: e1004287. doi: 10.1371/journal.pgen.1004287
|
| [19] |
Andrade MA, Chacon P, Merelo JJ, et al. (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng 6: 383-90. doi: 10.1093/protein/6.4.383
|
| [20] |
Petoukhov MV, Franke D, Shkumatov AV, et al. (2012) New developments in the program package for small-angle scattering data analysis. J Appl Crystallogr 45: 342-350. doi: 10.1107/S0021889812007662
|
| [21] |
Konarev PV, Svergun DI (2015) A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCrJ 2: 352-60. doi: 10.1107/S2052252515005163
|
| [22] |
Chacon P, Wriggers W (2002) Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol 317: 375-84. doi: 10.1006/jmbi.2002.5438
|
| [23] |
Wriggers W (2010) Using Situs for the integration of multi-resolution structures. Biophys Rev 2: 21-27. doi: 10.1007/s12551-009-0026-3
|
| [24] | Li X, Romero P, Rani M, et al. (1999) Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform Ser Workshop Genome Inform 10: 30-40. |
| [25] | Romero P, Obradovic Z, Dunker K (1997) Sequence Data Analysis for Long Disordered Regions Prediction in the Calcineurin Family. Genome Inform Ser Workshop Genome Inform 8: 110-124. |
| [26] | Romero P, Obradovic Z, Li X, et al. (2001) Sequence complexity of disordered protein. Proteins 42: 38-48. |
| [27] |
Borgstahl GE (2007) How to use dynamic light scattering to improve the likelihood of growing macromolecular crystals. Methods Mol Biol 363: 109-29. doi: 10.1007/978-1-59745-209-0_6
|
| [28] |
Garnier J, Gibrat JF, Robson B (1996) GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol 266: 540-53. doi: 10.1016/S0076-6879(96)66034-0
|
| [29] | Putnam CD, Hammel M, Hura GL, et al. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40: 191-285. |
| [30] |
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. doi: 10.1186/1471-2105-9-40
|
| [31] |
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725-38. doi: 10.1038/nprot.2010.5
|
| [32] | Yang J, Yan R, Roy A, et al. (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12: 7-8. |
| [33] |
Yang J, Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43: W174-81. doi: 10.1093/nar/gkv342
|
| [34] |
Buchan DW, Minneci F, Nugent TC, et al. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 41: W349-57. doi: 10.1093/nar/gkt381
|
| [35] |
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292: 195-202. doi: 10.1006/jmbi.1999.3091
|
| [36] |
Xu D, Jaroszewski L, Li Z, et al. (2014) FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics 30: 660-7. doi: 10.1093/bioinformatics/btt578
|
| [37] | Xu Y, Xu D (2000) Protein threading using PROSPECT: design and evaluation. Proteins 40: 343-54. |
| [38] |
Roy A, Yang J, Zhang Y (2012) COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40: W471-7. doi: 10.1093/nar/gks372
|
| [39] |
Svergun DI, Barberato C, Koch MHJ (1995) CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates J Appl Cryst 28: 768-773. doi: 10.1107/S0021889895007047
|
| [40] |
Zanier K, Charbonnier S, Sidi AO, et al. (2013) Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 339: 694-8. doi: 10.1126/science.1229934
|
| [41] |
Simister PC, Schaper F, O'Reilly N, et al. (2011) Self-organization and regulation of intrinsically disordered proteins with folded N-termini. PLoS Biol 9: e1000591. doi: 10.1371/journal.pbio.1000591
|
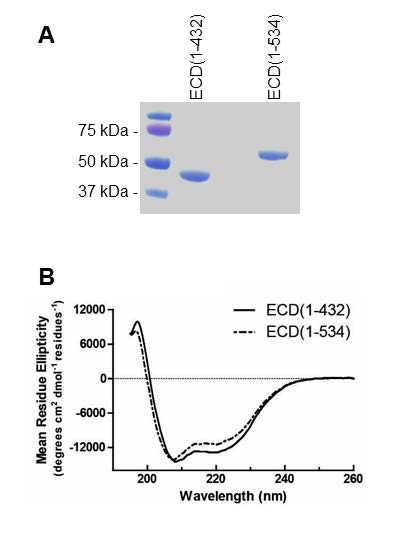
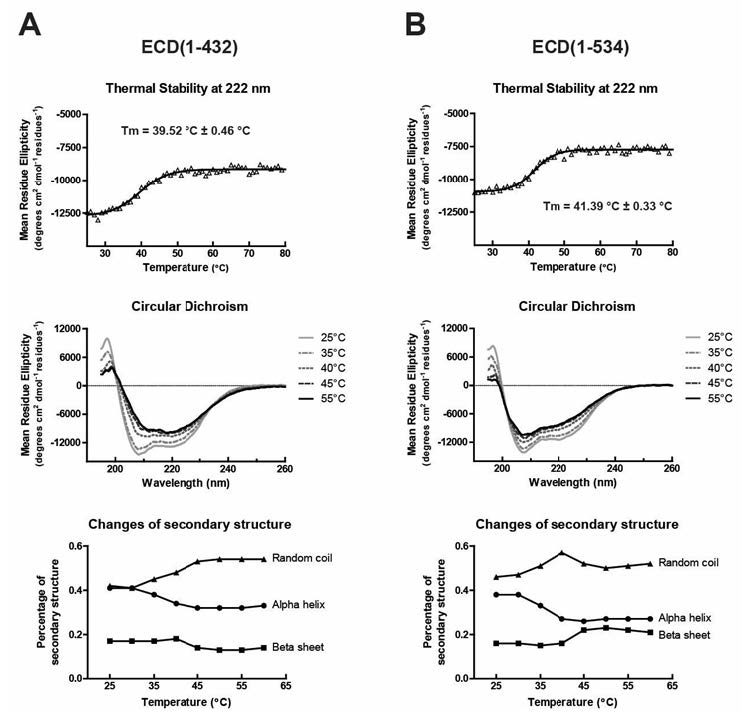
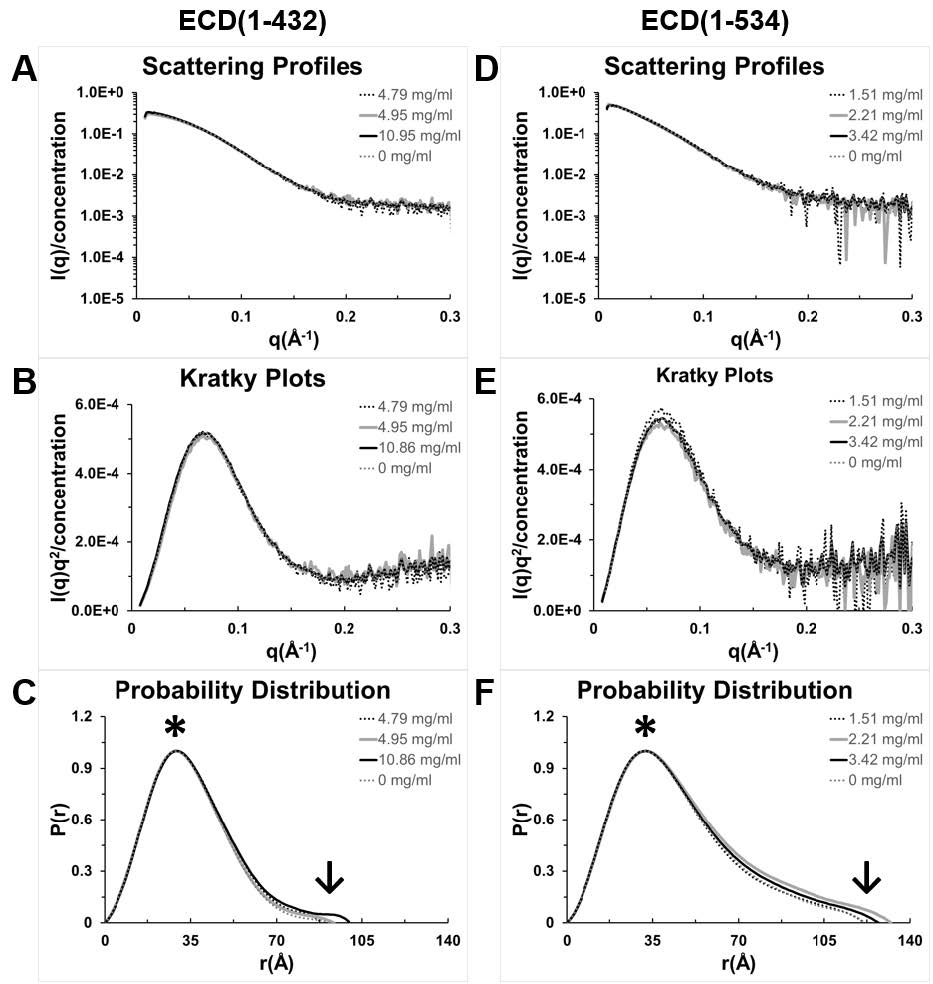
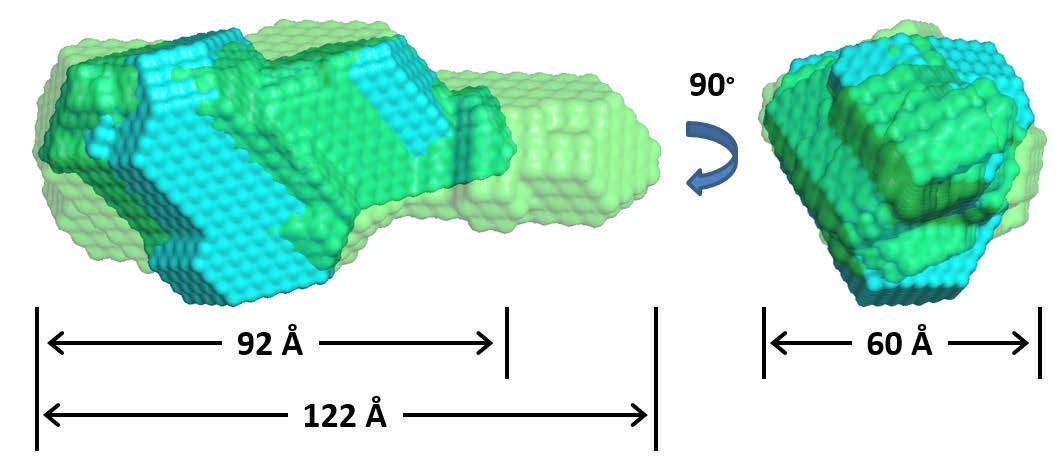

Figures(8) / Tables(1)

Riyaz A. Mir, Jeff Lovelace, Nicholas P. Schafer, Peter D. Simone, Admir Kellezi, Carol Kolar, Gaelle Spagnol, Paul L. Sorgen, Hamid Band, Vimla Band, Gloria E. O. Borgstahl. Biophysical characterization and modeling of human Ecdysoneless (ECD) protein supports a scaffolding function[J]. AIMS Biophysics, 2016, 3(1): 195-210. doi: 10.3934/biophy.2016.1.195







 DownLoad:
DownLoad: