Citation: T. Virmani, F. J. Urbano, V. Bisagno, E. Garcia-Rill. The pedunculopontine nucleus: From posture and locomotion to neuroepigenetics[J]. AIMS Neuroscience, 2019, 6(4): 219-230. doi: 10.3934/Neuroscience.2019.4.219

| [1] |
Garcia-Rill E (1986) The basal ganglia and the locomotor regions. Brain Res 11: 47–63. doi: 10.1016/0165-0173(86)90009-3

|
| [2] |
Garcia-Rill E (1991) The pedunculopontine nucleus. Prog Neurobiol 36: 363–389. doi: 10.1016/0301-0082(91)90016-T
|
| [3] |
Aziz TZ, Davies J, Stein J, et al. (1998) The role of descending basal ganglia connections to the brain stem in Parkinsonian akinesia. Brit J Neurosurg 12: 245–249. doi: 10.1080/02688699845078
|
| [4] | Garcia-Rill E, Mahaffey S, Hyde JR, et al. (2019) Bottom-up gamma maintenance in various disorders. In: Neurobiology of Disease: Pedunculopontine Nucleus Deep Brain Stimulation. Neurobiol Dis 128: 31–39. |
| [5] |
Ferraye MU, Debu B, Fraix V, et al. (2010) Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson's disease. Brain 133: 205–214. doi: 10.1093/brain/awp229
|
| [6] |
Mazzone P, Sposato S, Insola A, et al. (2008) Stereotactic surgery of nucleus tegmenti pedunculopontine [corrected]. Brit J Neurosurg 22: S33–S40. doi: 10.1080/02688690802448327
|
| [7] |
Moro E, Hamani C, Poon YY, et al. (2010) Unilateral pedunculopontine stimulation improves falls in Parkinson's disease. Brain 133: 215–224. doi: 10.1093/brain/awp261
|
| [8] |
Thevanasathan W, Silburn PA, Brooker H, et al. (2010) The impact of low-frequency stimulation of the pedunculopontine nucleus region on reaction time in Parkinsonism. J Neurol Neurosurg Psychiat 81: 1099–1104. doi: 10.1136/jnnp.2009.189324
|
| [9] |
Thevanasathan W, Cole MH, Grapel Cl, et al. (2012) A spatiotemporal analysis of gait freezing and the impact of pedunculopontine nucleus stimulation. Brain 135: 1446–1454. doi: 10.1093/brain/aws039
|
| [10] |
Vitale F, Capozzo A, Mazzone P, et al. (2019) Nweurophysiology of the pedunculopontine tegmental nucleus. Neurobiol Dis 128: 19–30. doi: 10.1016/j.nbd.2018.03.004
|
| [11] |
Costa A, Carlesimo GA, Caltagirone C, et al. (2010) Effects of deep brain stimulation of the peduncolopontine area on working memory tasks in patients with Parkinson's disease. Parkinsonism Relat Disord 16: 64–67. doi: 10.1016/j.parkreldis.2009.05.009
|
| [12] |
Lim AS, Moro E, Lozano A, et al. (2009) Selective enhancement of rapid eye movement sleep by deep brain stimulation of the human pons. Ann Neurol 66: 110–114. doi: 10.1002/ana.21631
|
| [13] |
Peppe A, Pierantozzi M, Baiamonte V, et al. (2012) Deep brain stimulation of pedunculopontine tegmental nucleus: role in sleep modulation in advanced Parkinson disease patients: one-year follow-up. Sleep 35: 1637–1642. doi: 10.5665/sleep.2234
|
| [14] |
Zanini S, Moschella V, Stefani A, et al. (2009) Grammar improvement following deep brain stimulation of the subthalamic and the pedunculopontine nuclei in advanced Parkinson's disease: a pilot study. Parkinsonism Relat Disord 15: 606–609. doi: 10.1016/j.parkreldis.2008.12.003
|
| [15] |
Stefani A, Pierantozzi M, Ceravolo R, et al. (2010) Deep brain stimulation of pedunculopontine tegmental nucleus (PPTg) promotes cognitive and metabolic changes: A target-specific effect or response to a low-frequency pattern of stimulation? Clin EEG Neurosci 41: 82–86. doi: 10.1177/155005941004100207
|
| [16] | Fahn S (1995) The freezing phenomenon in parkinsonism. Adv Neurol 67: 53–63. |
| [17] |
Schaafsma JD, Balash Y, Gurevich T, et al. (2003) Characterization of freezing of gait subtypes and the response of each to levodopa in Parkinson's disease. Eur J Neurol 10: 391–398. doi: 10.1046/j.1468-1331.2003.00611.x
|
| [18] |
Okuma Y, Silva de Lima AL, Fukae J, et al. (2018) A prospective study of falls in relation to freezing of gait and response fluctuations in Parkinson's disease. Parkinsonism Relat Disord 46: 30–35. doi: 10.1016/j.parkreldis.2017.10.013
|
| [19] |
Bloem BR, Hausdorff JM, Visser JE, et al. (2004) Falls and freezing of gait in Parkinson's disease: A review of two interconnected, episodic phenomena. Mov Disord 19: 871–884. doi: 10.1002/mds.20115
|
| [20] |
Johnell O, Melton LJ 3rd, Atkinson EJ, et al. (1992) Fracture risk in patients with parkinsonism: A population-based study in Olmsted County, Minnesota. Age Ageing 21: 32–38. doi: 10.1093/ageing/21.1.32
|
| [21] |
Adkin AL, Frank JS, Jog MS (2003) Fear of falling and postural control in Parkinson's disease. Mov Disord 18: 496–502. doi: 10.1002/mds.10396
|
| [22] |
Walton CC, Shine JM, Hall JM, et al. (2015) The major impact of freezing of gait on quality of life in Parkinson's disease. J Neurol 262: 108–115. doi: 10.1007/s00415-014-7524-3
|
| [23] |
Giladi N, McDermott MP, Fahn S, et al. (2001) Freezing of gait in PD: prospective assessment in the DATATOP cohort. Neurology 56: 1712–1721. doi: 10.1212/WNL.56.12.1712
|
| [24] |
Virmani T, Moskowitz CB, Vonsattel JP, et al. (2015) Clinicopathological characteristics of freezing of gait in autopsy-confirmed Parkinson's disease. Mov Disord 30: 1874–1884. doi: 10.1002/mds.26346
|
| [25] |
Espay AJ, Fasano A, van Nuenen BF, et al. (2012) "On" state freezing of gait in Parkinson disease: A paradoxical levodopa-induced complication. Neurology 78: 454–457. doi: 10.1212/WNL.0b013e3182477ec0
|
| [26] |
Nieuwboer A, Giladi N (2013) Characterizing freezing of gait in Parkinson's disease: models of an episodic phenomenon. Mov Disord 28: 1509–1519. doi: 10.1002/mds.25683
|
| [27] | Plotnik M, Giladi N, Hausdorff JM (2012) Is freezing of gait in Parkinson's disease a result of multiple gait impairments? Implications for treatment. Parkinsons Dis 2012: 459321. |
| [28] |
Lewis SJ, Barker RA (2009) A pathophysiological model of freezing of gait in Parkinson's disease. Parkinsonism Relat Disord 15: 333–338. doi: 10.1016/j.parkreldis.2008.08.006
|
| [29] | Vandenbossche J, Deroost N, Soetens E, et al. (2012) Freezing of gait in Parkinson's disease: disturbances in automaticity and control. Front Hum Neurosci 6: 356. |
| [30] |
Jacobs JV, Nutt JG, Carlson-Kuhta P, et al. (2009) Knee trembling during freezing of gait represents multiple anticipatory postural adjustments. Exp Neurol 215: 334–341. doi: 10.1016/j.expneurol.2008.10.019
|
| [31] |
Shah J, Pillai L, Williams DK, et al. (2018) Increased foot strike variability in Parkinson's disease patients with freezing of gait. Parkinsonism Relat Disord 53: 58–63. doi: 10.1016/j.parkreldis.2018.04.032
|
| [32] |
Virmani T, Pillai L, Glover A, et al. (2018) Impaired step-length setting prior to turning in Parkinson's disease patients with freezing of gait. Mov Disord 33: 1823–1825. doi: 10.1002/mds.27499
|
| [33] |
Nantel J, McDonald JC, Tan S, et al. (2012) Deficits in visuospatial processing contribute to quantitative measures of freezing of gait in Parkinson's disease. Neuroscience 221: 151–156. doi: 10.1016/j.neuroscience.2012.07.007
|
| [34] |
Amboni M, Cozzolino A, Longo K, et al. (2008) Freezing of gait and executive functions in patients with Parkinson's disease. Mov Disord 23: 395–400. doi: 10.1002/mds.21850
|
| [35] |
Braak H, Del Tredici K, Rub U, et al. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197–211. doi: 10.1016/S0197-4580(02)00065-9
|
| [36] | Garcia-Rill E (2015) Waking and the Reticular Activating System in Health and Disease, New York: Elsevier, 313. |
| [37] |
Moruzzi G, Magoun HW (1949) Brain stem reticular formation and activation of the EEG. Electroenceph Clin Neurophysiol 1: 455–473. doi: 10.1016/0013-4694(49)90219-9
|
| [38] | Steriade M (1999) Cellular substrates of oscillations in corticothalamic systems during states of vigilance, In: Lydic R, Baghdoyan HA, Editors, Handbook of Behavioral State Control. Cellular and molecular mechanisms, New York: CRC Press, 327–347. |
| [39] | Datta S, Siwek DF (2002) Single cell activity patterns of pedunculopontine tegmentum neurons across the sleep-wake cycle in the freely moving rats. J Neurosci Res 70: 79–82. |
| [40] | Shik ML, Severin FV, Orlovskii GN (1966) Control of walking and running by means of electric stimulation of the midbrain. Biofizika 11: 659–666. |
| [41] |
Garcia-Rill E, Houser C, Skinner RD, et al. (1987) Locomotion-inducing sites in the vicinity of the pedunculopontine nucleus. Brain Res Bull 18: 731–738. doi: 10.1016/0361-9230(87)90208-5
|
| [42] | Garcia-Rill E, Skinner D (1988) Modulation of rhythmic function in the posterior midbrain. Neurosci 17: 639–654. |
| [43] | Skinner RD, Garcia-Rill E (1990) Brainstem modulation of rhythmic functions and behaviors, In: Klemm WR, Vertes RP, Editors, Brainstem Mechanisms of Behavior, New York: John Wiley & Sons, 419–445. |
| [44] |
Garcia-Rill E, Skinner RD, Fitzgerald JA (1985) Chemical activation of the mesencephalic locomotor region. Brain Res 330: 43–54. doi: 10.1016/0006-8993(85)90006-X
|
| [45] | Garcia-Rill E, Skinner RD, Fitzgerald JA (1983) Activity in the mesencephalic locomotor region during locomotion. Exptl Neurol 82: 606–622. |
| [46] |
Kobayashi T, Good C, Biedermann J, et al (2004) Developmental changes in pedunculopontine nucleus (PPN) neurons. J Neurophysiol 91: 1470–1481. doi: 10.1152/jn.01024.2003
|
| [47] |
Reese NB, Garcia-Rill E, Skinner RD (1995) The pedunculopontine nucleus-auditory input, arousal and pathophysiology. Prog Neurobiol 47: 105–133. doi: 10.1016/0301-0082(95)00023-O
|
| [48] |
Wang HL, Morales M (2009) Pedunculopontine and laterodorsal tegmental nuclei contan distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci 29: 340–358. doi: 10.1111/j.1460-9568.2008.06576.x
|
| [49] |
Simon C, Kezunovic N, Ye M, et al. (2010) Gamma band unit activity and population responses in the pedunculopontine nucleus (PPN). J Neurophysiol 104: 463–474. doi: 10.1152/jn.00242.2010
|
| [50] |
Kezunovic N, Urbano FJ, Simon C, et al. (2011) Mechanism behind gamma band activity in the pedunculopontine nucleus (PPN). Eur J Neurosci 34: 404–415. doi: 10.1111/j.1460-9568.2011.07766.x
|
| [51] |
Luster B, D'Onofrio S, Urbano FJ, et al. (2015) High-Threshold Ca2+ channels behind gamma band activity in the pedunculopontine nucleus (PPN). Physiol Rep 3: e12431. doi: 10.14814/phy2.12431
|
| [52] |
Luster B, Urbano FJ, Garcia-Rill E (2016) Intracellular mechanisms modulating gamma band activity in the pedunculopontine nucleus (PPN). Physiol Rep 4: e12787. doi: 10.14814/phy2.12787
|
| [53] |
D'Onofrio S, Kezunovic N, Hyde JR, et al. (2015) Modulation of gamma oscillations in the pedunculopontine nucleus (PPN) by neuronal calcium sensor protein-1 (NCS-1): relevance to schizophrenia and bipolar disorder. J Neurophysiol 113: 709–719. doi: 10.1152/jn.00828.2014
|
| [54] |
D'Onofrio S, Urbano FJ, Mesias E, et al. (2016) Lithium decreases the effects of neuronal calcium sensor protein 1 in pedunculopontine neurons. Physiol Rep 4: e12740. doi: 10.14814/phy2.12740
|
| [55] |
Garcia-Rill E, Kezunovic N, D'Onofrio S, et al. (2014) Gamma band activity in the RAS- intracellular mechanisms. Exptl Brain Res 232: 1509–1522. doi: 10.1007/s00221-013-3794-8
|
| [56] |
Lai Y-Y, Siegel JM (1991) Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci 11: 2931–2937. doi: 10.1523/JNEUROSCI.11-09-02931.1991
|
| [57] |
Takakusaki K, Habaguchi T, Ohtinata-Sugimoto J, et al. (2003) Basal ganglia efferents to the brainstem centers controlling postural muscle tone and locomotion: A new concept for understanding motor disorders in basal ganglia dysfunction. Neuroscience 119: 293–308. doi: 10.1016/S0306-4522(03)00095-2
|
| [58] |
Ricciardi L, Sarchioto M, Morgante F (2019) Role of pedunculopontine nucleus in sleep-wake cycle and cognition in humans: A systematic review of DBS studies. Neurobiol Dis 128: 53–58. doi: 10.1016/j.nbd.2019.01.022
|
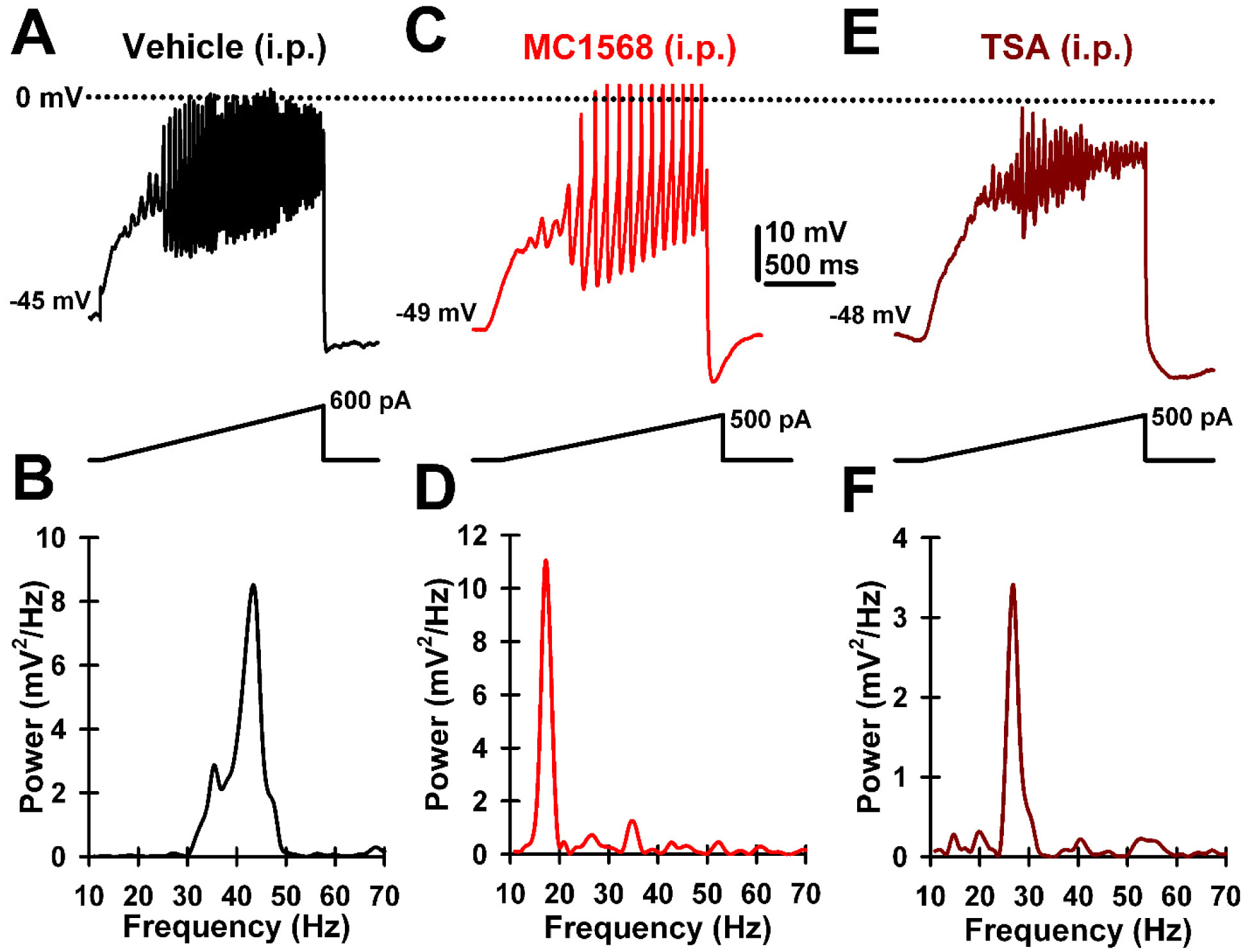
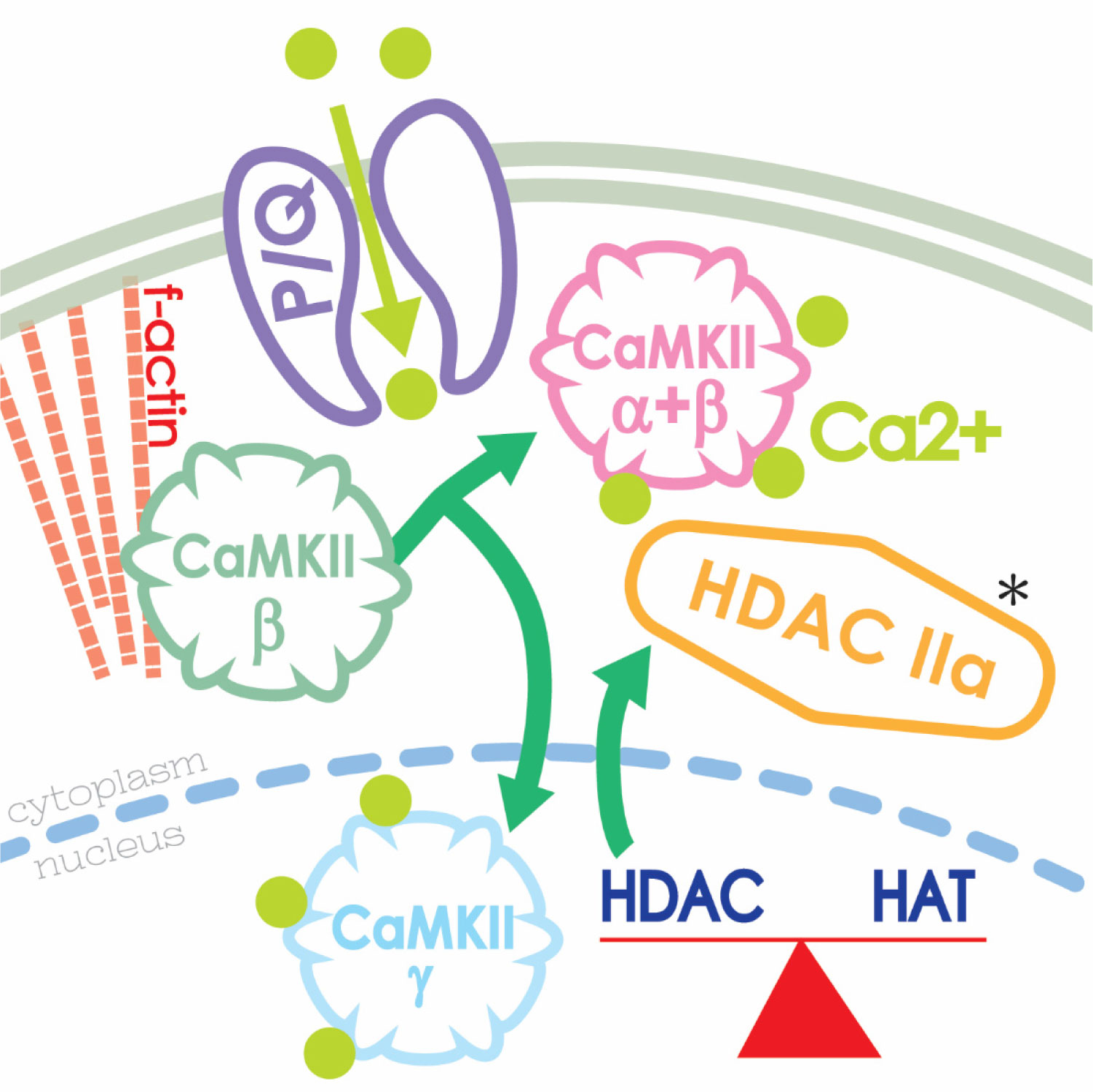
| [59] | Urbano FJ, Bisagno V, Mahaffey S, et al. (2018) Class II histone deacetylases require P/Q-type Ca2+ channels and CaMKII to maintain gamma oscillations in the pedunculopontine nucleus. Nature Sci Rep 8: 13156. |
| [60] | Urbano FJ, Bisagno V, Garcia-Rill E (2019) Neuroepigenetic control of gamma oscillations in the pedunculopontine nucleus: From HDACs to F-actin. Abstract #P-08-0375. International Brain Research Organization (IBRO) Meeting, Daegu, Korea. |
| [61] |
Garcia-Rill E, Tackett AJ, Byrum SD, et al. (2019) Local and relayed effects of deep brain stimulation of the pedunculopontine nucleus. Brain Sci 9: E64. doi: 10.3390/brainsci9030064
|
| [62] |
Byrum S, Washam C, Tackett A, et al. (2019) Proteomic measures of gamma oscillations. Heliyon 5: e02265. doi: 10.1016/j.heliyon.2019.e02265
|
| [63] |
Garcia-Rill E, Charlesworth A, Heister D, et al. (2008) The developmental decrease in REM sleep: The role of transmitters and electrical coupling. Sleep 31: 673–690. doi: 10.1093/sleep/31.5.673
|
| [64] |
Choudhary C, Kumar C, Gnad F, et al. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834–840. doi: 10.1126/science.1175371
|
Figures(3)

T. Virmani, F. J. Urbano, V. Bisagno, E. Garcia-Rill. The pedunculopontine nucleus: From posture and locomotion to neuroepigenetics[J]. AIMS Neuroscience, 2019, 6(4): 219-230. doi: 10.3934/Neuroscience.2019.4.219







 DownLoad:
DownLoad: