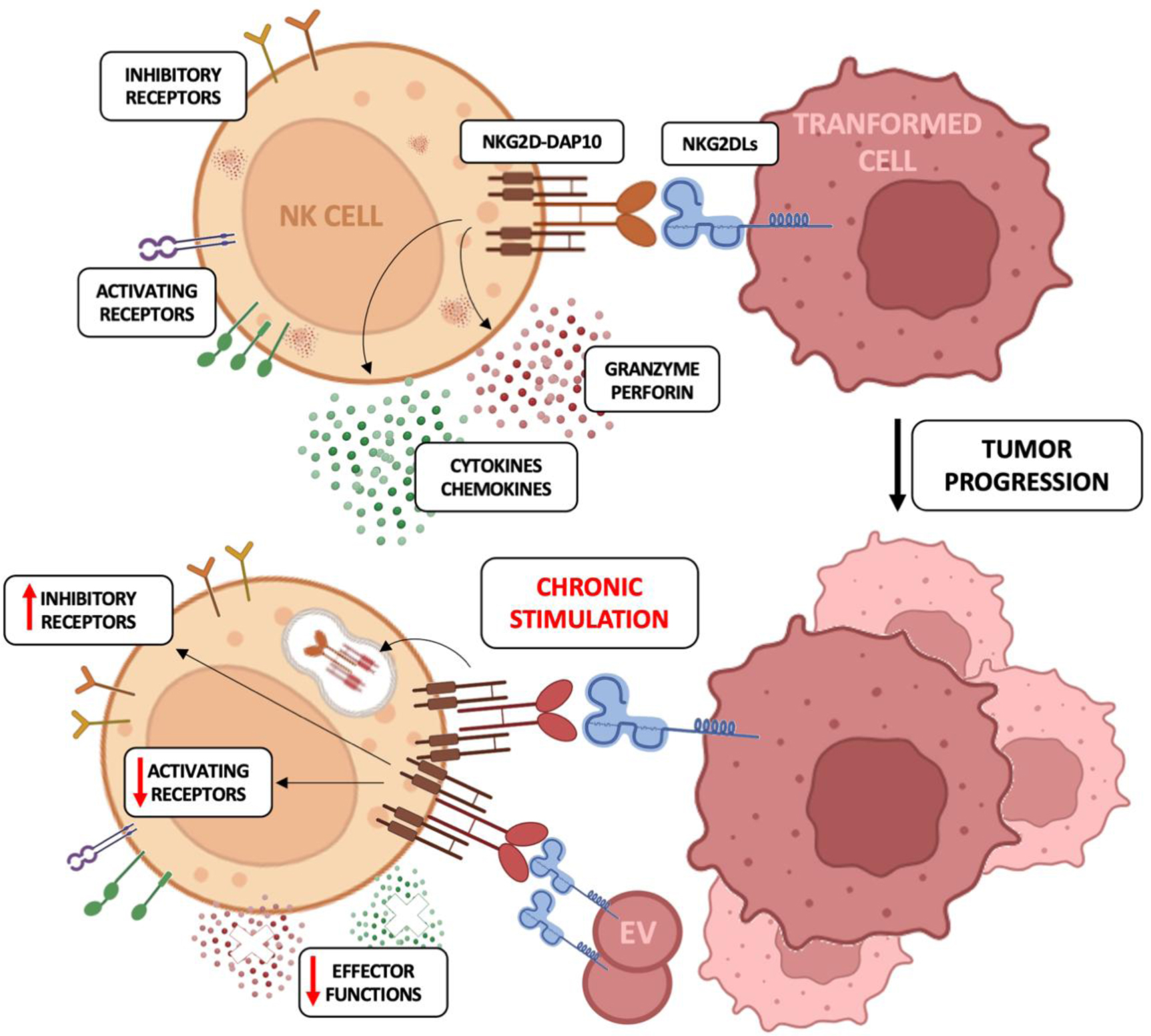
Natural killer (NK) cells are cytotoxic innate lymphocytes that represent the first line of defense against pathogen infections and tumor growth thanks to their ability to kill cancerous or infected cells and release pro-inflammatory cytokines. NK cell activation is regulated by the expression of a wide array of inhibitory receptors for MHC-I molecules and activating receptors, including NKG2D, which recognize self-ligands upregulated on stressed and damaged cells. Even though the expression of NKG2D ligands (NKG2DL) flags the target cells for NK cell-mediated elimination, a persistent interaction of NKG2D with its ligands promotes receptor downregulation, mainly through the internalization and lysosomal degradation of NKG2D/NKG2DL complexes, thus leading to an exhausted phenotype. On both human and murine NK cells, this phenotype is characterized by the down-modulation of the cytolytic machinery and the upregulation of inhibitory receptors.
In this review, we discuss the current knowledge on the contribution of the NKG2D/NKG2DL axis in both NK cell-mediated clearance of infected and transformed cells and in the dysregulation of NK cell activity due to chronic exposure to NKG2DLs during tumorigenesis.
Citation: Caterina Marangio, Rosa Molfetta, Erisa Putro, Alessia Carnevale, Rossella Paolini. Exploring the dynamic of NKG2D/NKG2DL axis: A central regulator of NK cell functions[J]. AIMS Allergy and Immunology, 2025, 9(2): 70-88. doi: 10.3934/Allergy.2025005
Natural killer (NK) cells are cytotoxic innate lymphocytes that represent the first line of defense against pathogen infections and tumor growth thanks to their ability to kill cancerous or infected cells and release pro-inflammatory cytokines. NK cell activation is regulated by the expression of a wide array of inhibitory receptors for MHC-I molecules and activating receptors, including NKG2D, which recognize self-ligands upregulated on stressed and damaged cells. Even though the expression of NKG2D ligands (NKG2DL) flags the target cells for NK cell-mediated elimination, a persistent interaction of NKG2D with its ligands promotes receptor downregulation, mainly through the internalization and lysosomal degradation of NKG2D/NKG2DL complexes, thus leading to an exhausted phenotype. On both human and murine NK cells, this phenotype is characterized by the down-modulation of the cytolytic machinery and the upregulation of inhibitory receptors.
In this review, we discuss the current knowledge on the contribution of the NKG2D/NKG2DL axis in both NK cell-mediated clearance of infected and transformed cells and in the dysregulation of NK cell activity due to chronic exposure to NKG2DLs during tumorigenesis.

| [1] |
Lanier LL (2008) Evolutionary struggles between NK cells and viruses. Nat Rev Immunol 8: 259-268. https://doi.org/10.1038/nri2276 
|
| [2] |
Long EO, Kim HS, Liu D, et al. (2013) Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu Rev Immunol 31: 227-258. https://doi.org/10.1146/annurev-immunol-020711-075005
|
| [3] |
Chiossone L, Dumas PY, Vienne M, et al. (2018) Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol 18: 671-688. https://doi.org/10.1038/s41577-018-0061-z
|
| [4] |
Ljunggren HG, Kärre K (1990) In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 11: 237-244. https://doi.org/10.1016/0167-5699(90)90097-s
|
| [5] |
Guia S, Fenis A, Vivier E, et al. (2018) Activating and inhibitory receptors expressed on innate lymphoid cells. Semin Immunopathol 40: 331-341. https://doi.org/10.1007/s00281-018-0685-x
|
| [6] |
Champsaur M, Lanier LL (2010) Effect of NKG2D ligand expression on host immune responses. Immunol Rev 235: 267-285. https://doi.org/10.1111/j.0105-2896.2010.00893.x
|
| [7] |
Raulet DH, Gasser S, Gowen BG, et al. (2013) Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 31: 413-441. https://doi.org/10.1146/annurev-immunol-032712-095951
|
| [8] |
Cerboni C, Fionda C, Soriani A, et al. (2014) The DNA Damage Response: A common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol 4: 508. https://doi.org/10.3389/fimmu.2013.00508
|
| [9] |
Lanier LL (2015) NKG2D receptor and its ligands in host defense. Cancer Immunol Res 3: 575-582. https://doi.org/10.1158/2326-6066.CIR-15-0098
|
| [10] |
Martinet L, Smyth MJ (2015) Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 15: 243-254. https://doi.org/10.1038/nri3799
|
| [11] |
Zingoni A, Molfetta R, Fionda C, et al. (2018) NKG2D and its ligands: “One for All, All for One”. Front Immunol 9: 476. https://doi.org/10.3389/fimmu.2018.00476
|
| [12] |
Barrow AD, Martin CJ, Colonna M (2019) The natural cytotoxicity receptors in health and disease. Front Immunol 10: 909. https://doi.org/10.3389/fimmu.2019.00909
|
| [13] |
Fuchs A, Colonna M (2006) The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol 16: 359-366. https://doi.org/10.1016/j.semcancer.2006.07.002
|
| [14] | González S, Groh V, Spies T (2006) Immunobiology of human NKG2D and its ligands. Curr Topics Microbiol Immunol 298: 121-138. https://doi.org/10.1007/3-540-27743-9_6 |
| [15] |
Fernández-Messina L, Reyburn HT, Valés-Gómez M (2012) Human NKG2D-ligands: Cell biology strategies to ensure immune recognition. Front Immunol 3: 299. https://doi.org/10.3389/fimmu.2012.00299
|
| [16] |
Ullrich E, Koch J, Cerwenka A, et al. (2013) New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2: e26097. https://doi.org/10.4161/onci.26097
|
| [17] |
Glienke J, Sobanov Y, Brostjan C, et al. (1998) The genomic organization of NKG2C, E, F, and D receptor genes in the human natural killer gene complex. Immunogenetics 48: 163-173. https://doi.org/10.1007/s002510050420
|
| [18] |
Basílio-Queirós D, Mischak-Weissinger E (2023) Natural killer cells- from innate cells to the discovery of adaptability. Front Immunol 14: 1172437. https://doi.org/10.3389/fimmu.2023.1172437
|
| [19] |
Huntington ND, Vosshenrich CA, Di Santo JP (2007) Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol 7: 703-714. https://doi.org/10.1038/nri2154
|
| [20] |
Zafirova B, Mandarić S, Antulov R, et al. (2009) Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity 31: 270-282. https://doi.org/10.1016/j.immuni.2009.06.017
|
| [21] |
Zafirova B, Wensveen FM, Gulin M, et al. (2011) Regulation of immune cell function and differentiation by the NKG2D receptor. Cell Mol Life Sci 68: 3519-3529. https://doi.org/10.1007/s00018-011-0797-0
|
| [22] |
Sheppard S, Triulzi C, Ardolino M, et al. (2013) Characterization of a novel NKG2D and NKp46 double-mutant mouse reveals subtle variations in the NK cell repertoire. Blood 121: 5025-5033. https://doi.org/10.1182/blood-2012-12-471607
|
| [23] |
Guerra N, Tan YX, Joncker NT, et al. (2008) NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28: 571-580. https://doi.org/10.1016/j.immuni.2008.02.016
|
| [24] |
Charpak-Amikam Y, Kournos M, Kotzur R, et al. (2024) The activating receptor NKG2D is an anti-fungal pattern recognition receptor. Nat Commun 15: 8664. https://doi.org/10.1038/s41467-024-52913-2
|
| [25] |
Molfetta R, Quatrini L, Zitti B, et al. (2016) Regulation of NKG2D expression and signaling by endocytosis. Trends Immunol 37: 790-802. https://doi.org/10.1016/j.it.2016.08.015
|
| [26] |
Wensveen FM, Jelenčić V, Polić B (2018) NKG2D: A master regulator of immune cell responsiveness. Front Immunol 9: 441. https://doi.org/10.3389/fimmu.2018.00441
|
| [27] |
Garrity D, Call ME, Feng J, et al. (2005) The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci U S A 102: 7641-7646. https://doi.org/10.1073/pnas.0502439102
|
| [28] |
Bauer S, Groh V, Wu J, et al. (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285: 727-729. https://doi.org/10.1126/science.285.5428.727
|
| [29] |
Wu J, Song Y, Bakker AB, et al. (1999) An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285: 730-732. https://doi.org/10.1126/science.285.5428.730
|
| [30] |
Billadeau DD, Upshaw JL, Schoon RA, et al. (2003) NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol 4: 557-564. https://doi.org/10.1038/ni929
|
| [31] |
Upshaw JL, Arneson LN, Schoon RA, et al. (2006) NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol 7: 524-532. https://doi.org/10.1038/ni1325
|
| [32] |
Diefenbach A, Tomasello E, Lucas M, et al. (2002) Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol 3: 1142-1149. https://doi.org/10.1038/ni858
|
| [33] |
Gilfillan S, Ho EL, Cella M, et al. (2002) NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nature Immunol 3: 1150-1155. https://doi.org/10.1038/ni857
|
| [34] |
Cella M, Fujikawa K, Tassi I, et al. (2004) Differential requirements for Vav proteins in DAP10- and ITAM-mediated NK cell cytotoxicity. J Exp Med 200: 817-823. https://doi.org/10.1084/jem.20031847
|
| [35] |
Graham DB, Cella M, Giurisato E, et al. (2006) Vav1 controls DAP10-mediated natural cytotoxicity by regulating actin and microtubule dynamics. J Immunol 177: 2349-2355. https://doi.org/10.4049/jimmunol.177.4.2349
|
| [36] |
Vivier E, Tomasello E, Baratin M, et al. (2008) Functions of natural killer cells. Nat Immunol 9: 503-510. https://doi.org/10.1038/ni1582
|
| [37] |
Zompi S, Hamerman JA, Ogasawara K, et al. (2003) NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat Immunol 4: 565-572. https://doi.org/10.1038/ni930
|
| [38] |
Bryceson YT, March ME, Ljunggren HG, et al. (2006) Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107: 159-166. https://doi.org/10.1182/blood-2005-04-1351
|
| [39] |
Bryceson YT, Ljunggren HG, Long EO (2009) Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 114: 2657-2666. https://doi.org/10.1182/blood-2009-01-201632
|
| [40] |
Kim HS, Das A, Gross CC, et al. (2010) Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity 32: 175-186. https://doi.org/10.1016/j.immuni.2010.02.004
|
| [41] |
Ogasawara K, Hamerman JA, Hsin H, et al. (2003) Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity 18: 41-51. https://doi.org/10.1016/s1074-7613(02)00505-8
|
| [42] |
Quatrini L, Molfetta R, Zitti B, et al. (2015) Ubiquitin-dependent endocytosis of NKG2D-DAP10 receptor complexes activates signaling and functions in human NK cells. Sci Signaling 8: ra108. https://doi.org/10.1126/scisignal.aab2724
|
| [43] |
Roda-Navarro P, Reyburn HT (2009) The traffic of the NKG2D/Dap10 receptor complex during natural killer (NK) cell activation. Journal Biol Chem 284: 16463-16472. https://doi.org/10.1074/jbc.M808561200
|
| [44] |
Molfetta R, Quatrini L, Capuano C, et al. (2014) c-Cbl regulates MICA- but not ULBP2-induced NKG2D down-modulation in human NK cells. Eur J Immunol 44: 2761-2770. https://doi.org/10.1002/eji.201444512
|
| [45] |
Oppenheim DE, Roberts SJ, Clarke SL, et al. (2005) Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol 6: 928-937. https://doi.org/10.1038/ni1239
|
| [46] |
Coudert JD, Scarpellino L, Gros F, et al. (2008) Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 111: 3571-3578. https://doi.org/10.1182/blood-2007-07-100057
|
| [47] |
Molfetta R, Zingoni A, Santoni A, et al. (2019) Post-translational mechanisms regulating NK cell activating receptors and their ligands in cancer: Potential targets for therapeutic intervention. Front Immunol 10: 2557. https://doi.org/10.3389/fimmu.2019.02557
|
| [48] |
Gasser S, Orsulic S, Brown EJ, et al. (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436: 1186-1190. https://doi.org/10.1038/nature03884
|
| [49] |
Soriani A, Zingoni A, Cerboni C, et al. (2009) ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113: 3503-3511. https://doi.org/10.1182/blood-2008-08-173914
|
| [50] |
Groh V, Bahram S, Bauer S, et al. (1996) Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A 93: 12445-12450. https://doi.org/10.1073/pnas.93.22.12445
|
| [51] |
Li H, Lakshmikanth T, Garofalo C, et al. (2011) Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle 10: 3346-3358. https://doi.org/10.4161/cc.10.19.17630
|
| [52] |
Textor S, Fiegler N, Arnold A, et al. (2011) Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res 71: 5998-6009. https://doi.org/10.1158/0008-5472.CAN-10-3211
|
| [53] |
Moretta L, Biassoni R, Bottino C, et al. (2002) Human NK cells and their receptors. Microbes Infect 4: 1539-1544. https://doi.org/10.1016/s1286-4579(02)00037-0
|
| [54] |
Gleimer M, Parham P (2003) Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity 19: 469-477. https://doi.org/10.1016/s1074-7613(03)00272-3
|
| [55] |
Baychelier F, Vieillard V (2013) The modulation of the cell-cycle: a sentinel to alert the NK cells of dangers. Front Immunol 4: 325. https://doi.org/10.3389/fimmu.2013.00325
|
| [56] |
Parsons MS, Richard J, Lee WS, et al. (2016) NKG2D Acts as a Co-Receptor for Natural Killer Cell-Mediated Anti-HIV-1 Antibody-Dependent Cellular Cytotoxicity. AIDS Res Human Retroviru 32: 1089-1096. https://doi.org/10.1089/AID.2016.0099
|
| [57] |
Ward J, Davis Z, DeHart J, et al. (2009) HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog 5: e1000613. https://doi.org/10.1371/journal.ppat.1000613
|
| [58] |
Richard J, Sindhu S, Pham TN, et al. (2010) HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115: 1354-1363. https://doi.org/10.1182/blood-2009-08-237370
|
| [59] |
Castillo JP, Frame FM, Rogoff HA, et al. (2005) Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J Virol 79: 11467-11475. https://doi.org/10.1128/JVI.79.17.11467-11475.2005
|
| [60] |
Xiaofei E, Pickering MT, Debatis M, et al. (2011) An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog 7: e1001342. https://doi.org/10.1371/journal.ppat.1001342
|
| [61] |
Pignoloni B, Fionda C, Dell'Oste V, et al. (2016) Distinct roles for human cytomegalovirus immediate early proteins IE1 and IE2 in the transcriptional regulation of MICA and PVR/CD155 expression. J Immunol 197: 4066-4078. https://doi.org/10.4049/jimmunol.1502527
|
| [62] |
Lodoen M, Ogasawara K, Hamerman JA, et al. (2003) NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med 197: 1245-1253. https://doi.org/10.1084/jem.20021973
|
| [63] |
Lodoen MB, Abenes G, Umamoto S, et al. (2004) The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med 200: 1075-1081. https://doi.org/10.1084/jem.20040583
|
| [64] |
Krmpotic A, Hasan M, Loewendorf A, et al. (2005) NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201: 211-220. https://doi.org/10.1084/jem.20041617
|
| [65] |
Hasan M, Krmpotic A, Ruzsics Z, et al. (2005) Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J Virol 79: 2920-2930. https://doi.org/10.1128/jvi.79.5.2920-2930.2005
|
| [66] |
Wilkinson GW, Tomasec P, Stanton RJ, et al. (2008) Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 41: 206-212. https://doi.org/10.1016/j.jcv.2007.10.027
|
| [67] |
Rossini G, Cerboni C, Santoni A, et al. (2012) Interplay between human cytomegalovirus and intrinsic/innate host responses: A complex bidirectional relationship. Mediators Inflammation 2012: 607276. https://doi.org/10.1155/2012/607276
|
| [68] |
Fielding CA, Aicheler R, Stanton RJ, W, et al. (2014) Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 10: e1004058. https://doi.org/10.1371/journal.ppat.1004058
|
| [69] |
Dunn C, Chalupny NJ, Sutherland CL, et al. (2003) Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 197: 1427-1439. https://doi.org/10.1084/jem.20022059
|
| [70] |
Welte SA, Sinzger C, Lutz SZ, et al. (2003) Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol 33: 194-203. https://doi.org/10.1002/immu.200390022
|
| [71] |
Müller S, Zocher G, Steinle A, et al. (2010) Structure of the HCMV UL16-MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PLoS Pathog 6: e1000723. https://doi.org/10.1371/journal.ppat.1000723
|
| [72] |
Ashiru O, Bennett NJ, Boyle LH, et al. (2009) NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 83: 12345-12354. https://doi.org/10.1128/JVI.01175-09
|
| [73] |
Bennett NJ, Ashiru O, Morgan FJ, P, et al. (2010) Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J Immunol 185: 1093-1102. https://doi.org/10.4049/jimmunol.1000789
|
| [74] |
Cerboni C, Neri F, Casartelli N, et al. (2007) Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J General Virol 88: 242-250. https://doi.org/10.1099/vir.0.82125-0
|
| [75] |
Thomas M, Boname JM, Field S, et al. (2008) Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc Natl Acad Sci U S A 105: 1656-1661. https://doi.org/10.1073/pnas.0707883105
|
| [76] |
Zdrenghea MT, Telcian AG, Laza-Stanca V, et al. (2012) RSV infection modulates IL-15 production and MICA levels in respiratory epithelial cells. Eur Respiratory J 39: 712-720. https://doi.org/10.1183/09031936.00099811
|
| [77] |
Reilly RB, Ramdour SK, Fuhlbrigge ME, et al. (2024) An altered natural killer cell immunophenotype characterizes clinically severe pediatric RSV infection. Sci Transl Med 16: eado6606. https://doi.org/10.1126/scitranslmed.ado6606
|
| [78] |
Fernández-Soto D, García-Jiménez ÁF, Casasnovas JM, et al. (2024) Elevated levels of cell-free NKG2D-ligands modulate NKG2D surface expression and compromise NK cell function in severe COVID-19 disease. Front Immunol 15: 1273942. https://doi.org/10.3389/fimmu.2024.1273942
|
| [79] |
Azzi T, Lünemann A, Murer A, et al. (2014) Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 124: 2533-2543. https://doi.org/10.1182/blood-2014-01-553024
|
| [80] |
Desimio MG, Covino DA, Cancrini C, et al. (2024) Entry into the lytic cycle exposes EBV-infected cells to NK cell killing via upregulation of the MICB ligand for NKG2D and activation of the CD56bright and NKG2A+KIR+CD56dim subsets. Front Immunol 15: 1467304. https://doi.org/10.3389/fimmu.2024.1467304
|
| [81] |
Pappworth IY, Wang EC, Rowe M (2007) The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol 81: 474-482. https://doi.org/10.1128/JVI.01777-06
|
| [82] |
Williams LR, Quinn LL, Rowe M, et al. (2015) Induction of the lytic cycle sensitizes epstein-barr virus-infected B cells to NK cell killing that is counteracted by virus-mediated NK cell evasion mechanisms in the late lytic cycle. J Virol 90: 947-958. https://doi.org/10.1128/JVI.01932-15
|
| [83] |
Desimio MG, Covino DA, Rivalta B, et al. (2023) The role of NK cells in EBV infection and related diseases: Current Understanding and hints for novel therapies. Cancers 15: 1914. https://doi.org/10.3390/cancers15061914
|
| [84] |
Lenart M, Kluczewska A, Szaflarska A, et al. (2021) Selective downregulation of natural killer activating receptors on NK cells and upregulation of PD-1 expression on T cells in children with severe and/or recurrent Herpes simplex virus infections. Immunobiology 226: 152097. https://doi.org/10.1016/j.imbio.2021.152097
|
| [85] |
Osman MS, van Eeden C, Cohen Tervaert JW (2020) Fatal COVID-19 infections: Is NK cell dysfunction a link with autoimmune HLH?. Autoimmun Rev 19: 102561. https://doi.org/10.1016/j.autrev.2020.102561
|
| [86] |
Song H, Park H, Kim J, et al. (2011) IDO metabolite produced by EBV-transformed B cells inhibits surface expression of NKG2D in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Immunol Lett 136: 187-193. https://doi.org/10.1016/j.imlet.2011.01.009
|
| [87] |
Doubrovina ES, Doubrovin MM, Vider E, et al. (2003) Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol 171: 6891-6899. https://doi.org/10.4049/jimmunol.171.12.6891
|
| [88] |
Wiemann K, Mittrücker HW, Feger U, et al. (2005) Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol 175: 720-729. https://doi.org/10.4049/jimmunol.175.2.720
|
| [89] |
Waldhauer I, Steinle A (2006) Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res 66: 2520-2526. https://doi.org/10.1158/0008-5472.CAN-05-2520
|
| [90] |
Clayton A, Mitchell JP, Court J, et al. (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180: 7249-7258. https://doi.org/10.4049/jimmunol.180.11.7249
|
| [91] |
Vulpis E, Loconte L, Peri A, et al. (2022) Impact on NK cell functions of acute versus chronic exposure to extracellular vesicle-associated MICA: Dual role in cancer immunosurveillance. J Extracell Vesicles 11: e12176. https://doi.org/10.1002/jev2.12176
|
| [92] |
Mamessier E, Sylvain A, Thibult ML, et al. (2011) Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest 121: 3609-3622. https://doi.org/10.1172/JCI45816
|
| [93] | Han B, Mao FY, Zhao YL, et al. (2018) Altered NKp30, NKp46, NKG2D, and DNAM-1 expression on circulating NK cells is associated with tumor progression in human gastric cancer. J Immunol Res 2018: 6248590. https://doi.org/10.1155/2018/6248590 |
| [94] |
Marcon F, Zuo J, Pearce H, et al. (2020) NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. Oncoimmunology 9: 1845424. https://doi.org/10.1080/2162402X.2020.1845424
|
| [95] |
von Lilienfeld-Toal M, Frank S, Leyendecker C, et al. (2010) Reduced immune effector cell NKG2D expression and increased levels of soluble NKG2D ligands in multiple myeloma may not be causally linked. Cancer Immunol Immunother 59: 829-839. https://doi.org/10.1007/s00262-009-0807-3
|
| [96] |
Hilpert J, Grosse-Hovest L, Grünebach F, et al. (2012) Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J Immunol 189: 1360-1371. https://doi.org/10.4049/jimmunol.1200796
|
| [97] |
Wiesmayr S, Webber SA, Macedo C, et al. (2012) Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol 42: 541-550. https://doi.org/10.1002/eji.201141832
|
| [98] |
Salih HR, Rammensee HG, Steinle A (2002) Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J Immunol 169: 4098-4102. https://doi.org/10.4049/jimmunol.169.8.4098
|
| [99] |
Cao W, Xi X, Hao Z, et al. (2007) RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J Biol Chem 282: 18922-18928. https://doi.org/10.1074/jbc.M702504200
|
| [100] |
Boutet P, Agüera-González S, Atkinson S, et al. (2009) Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol 182: 49-53. https://doi.org/10.4049/jimmunol.182.1.49
|
| [101] |
Ashiru O, Boutet P, Fernández-Messina L, et al. (2010) Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res 70: 481-489. https://doi.org/10.1158/0008-5472.CAN-09-1688
|
| [102] | Tamaki S, Kawakami M, Ishitani A, et al. (2010) Soluble MICB serum levels correlate with disease stage and survival rate in patients with oral squamous cell carcinoma. Anticancer Res 30: 4097-4101. |
| [103] |
Yamaguchi K, Chikumi H, Shimizu A, et al. (2012) Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer Sci 103: 1405-1413. https://doi.org/10.1111/j.1349-7006.2012.02330.x
|
| [104] |
Wu BJ, Li WP, Qian C, et al. (2013) Serum soluble MICB (sMICB) correlates with disease progression and survival in melanoma patients. Tumour Biol 34: 565-569. https://doi.org/10.1007/s13277-012-0582-1
|
| [105] |
Fernández-Messina L, Ashiru O, Boutet P, et al. (2010) Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J Biol Chem 285: 8543-8551. https://doi.org/10.1074/jbc.M109.045906
|
| [106] |
Gill S, Vasey AE, De Souza A, et al. (2012) Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood 119: 5758-5768. https://doi.org/10.1182/blood-2012-03-415364
|
| [107] |
Felices M, Lenvik AJ, McElmurry R, et al. (2018) Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 3: e96219. https://doi.org/10.1172/jci.insight.96219
|
| [108] |
Myers JA, Schirm D, Bendzick L, et al. (2022) Balanced engagement of activating and inhibitory receptors mitigates human NK cell exhaustion. JCI Insight 7: e150079. https://doi.org/10.1172/jci.insight.150079
|
| [109] |
Alvarez M, Simonetta F, Baker J, et al. (2019) Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight 5: e127729. https://doi.org/10.1172/jci.insight.127729
|
| [110] |
Milito ND, Zingoni A, Stabile H, et al. (2023) NKG2D engagement on human NK cells leads to DNAM-1 hypo-responsiveness through different converging mechanisms. Eur J Immunol 53: 2250198. https://doi.org/10.1002/eji.202250198
|
| [111] |
Hanaoka N, Jabri B, Dai Z, et al. (2010) NKG2D initiates caspase-mediated CD3zeta degradation and lymphocyte receptor impairments associated with human cancer and autoimmune disease. J Immunol 185: 5732-5742. https://doi.org/10.4049/jimmunol.1002092
|
| [112] |
Santiago V, Rezvani K, Sekine T, et al. (2018) Human NK Cells Develop an Exhaustion Phenotype During Polar Degranulation at the Aspergillus fumigatus Hyphal Synapse. Front Immunol 9: 2344. https://doi.org/10.3389/fimmu.2018.02344
|
| [113] |
Bär E, Whitney PG, Moor K, et al. (2014) IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity 40: 117-127. https://doi.org/10.1016/j.immuni.2013.12.002
|
Figures(1) / Tables(1)

Caterina Marangio, Rosa Molfetta, Erisa Putro, Alessia Carnevale, Rossella Paolini. Exploring the dynamic of NKG2D/NKG2DL axis: A central regulator of NK cell functions[J]. AIMS Allergy and Immunology, 2025, 9(2): 70-88. doi: 10.3934/Allergy.2025005







 DownLoad:
DownLoad: