Citation: Davide Sala, Andrea Giachetti, Antonio Rosato. Molecular dynamics simulations of metalloproteins: A folding study of rubredoxin from Pyrococcus furiosus[J]. AIMS Biophysics, 2018, 5(1): 77-96. doi: 10.3934/biophy.2018.1.77

| [1] |
Klepeis JL, Lindorff-Larsen K, Dror RO, et al. (2009) Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol 19: 120–127. doi: 10.1016/j.sbi.2009.03.004

|
| [2] |
Stone JE, Phillips JC, Freddolino PL, et al. (2007) Accelerating molecular modeling applications with graphics processors. J Comput Chem 28: 2618–2640. doi: 10.1002/jcc.20829
|
| [3] |
Perez A, Morrone JA, Simmerling C, et al. (2016) Advances in free-energy-based simulations of protein folding and ligand binding. Curr Opin Struct Biol 36: 25–31. doi: 10.1016/j.sbi.2015.12.002
|
| [4] |
Lane TJ, Shukla D, Beauchamp KA, et al. (2013) To milliseconds and beyond: Challenges in the simulation of protein folding. Curr Opin Struct Biol 23: 58–65. doi: 10.1016/j.sbi.2012.11.002
|
| [5] |
Freddolino PL, Harrison CB, Liu Y, et al. (2010) Challenges in protein-folding simulations. Nat Phys 6: 751–758. doi: 10.1038/nphys1713
|
| [6] |
Best RB (2012) Atomistic molecular simulations of protein folding. Curr Opin Struct Biol 22: 52–61. doi: 10.1016/j.sbi.2011.12.001
|
| [7] | Piana S, Lindorff-Larsen K, Shaw DE (2012) Protein folding kinetics and thermodynamics from atomistic simulation. P Natl Acad Sci USA 109: 17845–17850. |
| [8] |
Suárez E, Lettieri S, Zwier MC, et al. (2014) Simultaneous computation of dynamical and equilibrium information using a weighted ensemble of trajectories. J Chem Theory Comput 10: 2658–2667. doi: 10.1021/ct401065r
|
| [9] |
Pierce LCT, Salomon-Ferrer R, Augusto F. De Oliveira C, et al. (2012) Routine access to millisecond time scale events with accelerated molecular dynamics. J Chem Theory Comput 8: 2997–3002. doi: 10.1021/ct300284c
|
| [10] |
Kubelka J, Hofrichter J, Eaton WA (2004) The protein folding "speed limit." Curr Opin Struct Biol 14: 76–88. doi: 10.1016/j.sbi.2004.01.013
|
| [11] |
Lindorff-Larsen K, Piana S, Dror RO, et al. (2011) How fast-folding proteins fold. Science 334: 517–520. doi: 10.1126/science.1208351
|
| [12] | Putignano V, Rosato A, Banci L, et al. (2018) MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 41: 459–464. |
| [13] |
Li W, Wang J, Zhang J, et al. (2015) Molecular simulations of metal-coupled protein folding. Curr Opin Struct Biol 30: 25–31. doi: 10.1016/j.sbi.2014.11.006
|
| [14] |
Bentrop D, Bertini I, Iacoviello R, et al. (1999) Structural and dynamical properties of a partially unfolded Fe4S4 protein: Role of the cofactor in protein folding. Biochemistry 38: 4669–4680. doi: 10.1021/bi982647q
|
| [15] |
Blake PR, Summers MF, Park JB, et al. (1991) Determinants of protein hyperthermostability: purification and amino acid sequence of rubredoxin from the hyperthermophilic archaebacterium pyrococcus furiosus and secondary structure of the zinc adduct by NMR. Biochemistry 30: 10885–10895. doi: 10.1021/bi00109a012
|
| [16] | Prakash S, Sundd M, Guptasarma P (2014) The key to the extraordinary thermal stability of P. furiosus holo-rubredoxin: Iron binding-guided packing of a core aromatic cluster responsible for high kinetic stability of the native structure. PLoS One 9: e89703. |
| [17] |
Hernandez G, Jenney FE, Adams MW, et al. (2000) Millisecond time scale conformational flexibility in a hyperthermophile protein at ambient temperature. Proc Natl Acad Sci USA 97: 3166–3170. doi: 10.1073/pnas.97.7.3166
|
| [18] | Rader AJ (2010) Thermostability in rubredoxin and its relationship to mechanical rigidity. Phys Biol 7: 016002. |
| [19] |
Bonomi F, Iametti S, Ferranti P, et al. (2008) "Iron priming" guides folding of denatured aporubredoxins. J Biol Inorg Chem 13: 981–991. doi: 10.1007/s00775-008-0385-4
|
| [20] |
Zartler ER, Jenney FE, Terrell M, et al. (2001) Structural basis for thermostability in aporubredoxins from Pyrococcus furiosus and Clostridium pasteurianum. Biochemistry 40: 7279–7290. doi: 10.1021/bi0026831
|
| [21] |
Cavagnero S, Debe DA, Zhou ZH, et al. (1998) Kinetic role of electrostatic interactions in the unfolding of hyperthermophilic and mesophilic rubredoxins. Biochemistry 37: 3369–3376. doi: 10.1021/bi9721795
|
| [22] |
Strop P, Mayo SL (1999) Rubredoxin variant folds without iron. J Am Chem Soc 121: 2341–2345. doi: 10.1021/ja9834780
|
| [23] |
Hamelberg D, Mongan J, McCammon JA (2004) Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J Chem Phys 120: 11919–11929. doi: 10.1063/1.1755656
|
| [24] |
Doshi U, Hamelberg D (2015) Towards fast, rigorous and efficient conformational sampling of biomolecules: Advances in accelerated molecular dynamics. BBA-Gen Subjects 1850: 878–888. doi: 10.1016/j.bbagen.2014.08.003
|
| [25] |
Miao Y, Feixas F, Eun C (2015) Accelerated molecular dynamics simulations of protein folding. J Comput Chem 36: 1536–1549. doi: 10.1002/jcc.23964
|
| [26] | Case DA, Cerutti DS, Cheatham TE, et al. (2017) Amber 2017, University of California, San Francisco. |
| [27] |
Carvalho ATP, Teixeira AFS, Ramos MJ (2013) Parameters for molecular dynamics simulations of iron-sulfur proteins. J Comput Chem 34: 1540–1548. doi: 10.1002/jcc.23287
|
| [28] |
Bertini I, Case DA, Ferella L, et al. (2011) A grid-enabled web portal for NMR structure refinement with AMBER. Bioinformatics 27: 2384–2390. doi: 10.1093/bioinformatics/btr415
|
| [29] |
Wassenaar TA, van Dijk M, Loureiro-Ferreira N, et al. (2012) WeNMR: Structural biology on the grid. J Grid Comput 10: 743–767. doi: 10.1007/s10723-012-9246-z
|
| [30] |
Prompers JJ, Brüschweiler R, Bruschweiler R (2002) General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J Am Chem Soc 124: 4522–4534. doi: 10.1021/ja012750u
|
| [31] |
Korzhnev DM, Billeter M, Arseniev AS, et al. (2001) NMR studies of Brownian tumbling and internal motions in proteins. Prog Nucl Mag Res Sp 38: 197–266. doi: 10.1016/S0079-6565(00)00028-5
|
| [32] |
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. doi: 10.1002/bip.360221211
|
| [33] |
Rost B, Sander C (1993) Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol 232: 584–599. doi: 10.1006/jmbi.1993.1413
|
| [34] |
Li DW, Brüschweiler R (2012) PPM: A side-chain and backbone chemical shift predictor for the assessment of protein conformational ensembles. J Biomol NMR 54: 257–265. doi: 10.1007/s10858-012-9668-8
|
| [35] |
Hiller R, Zhou ZH, Adams MW, et al. (1997) Stability and dynamics in a hyperthermophilic protein with melting temperature close to 200 degrees C. Proc Natl Acad Sci USA 94: 11329–11332. doi: 10.1073/pnas.94.21.11329
|
| [36] |
Ishima R, Torchia DA (2000) Protein dynamics from NMR. Nat Struct Biol 7: 740–743. doi: 10.1038/78963
|
| [37] |
Jarymowycz VA, Stone MJ (2006) Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev 106: 1624–1671. doi: 10.1021/cr040421p
|
| [38] |
LeMaster DM (1999) NMR relaxation order parameter analysis of the dynamics of protein side chains. J Am Chem Soc 121: 1726–1742. doi: 10.1021/ja982988r
|
| [39] |
Ruschak AM, Kay LE (2010) Methyl groups as probes of supra-molecular structure, dynamics and function. J Biomol NMR 46: 75–87. doi: 10.1007/s10858-009-9376-1
|
| [40] |
Bougault CM, Eidsness MK, Prestegard JH (2003) Hydrogen bonds in rubredoxins from mesophilic and hyperthermophilic organisms. Biochemistry 42: 4357–4372. doi: 10.1021/bi027264d
|
| [41] |
Prestegard JH, Bougault CM, Kishore AI (2004) Residual dipolar couplings in structure determination of biomolecules. Chem Rev 104: 3519–3540. doi: 10.1021/cr030419i
|
| [42] |
Cho-Chung YS, Pitot HC (1968) Regulatory effects of nicotinamide on tryptophan pyrrolase synthesis in rat liver in vivo. Eur J Biochem 3: 401–406. doi: 10.1111/j.1432-1033.1967.tb19543.x
|
| [43] |
Blasie CA, Berg JM (2002) Structur e-based thermodynamic analysis of a coupled metal binding-protein folding reaction involving a zinc finger peptide. Biochemistry 41: 15068–15073. doi: 10.1021/bi026621h
|
| [44] |
Weinkam P, Romesberg FE, Wolynes PG (2009) Chemical frustration in the protein folding landscape: Grand canonical ensemble simulations of cytochrome c. Biochemistry 48: 2394–2402. doi: 10.1021/bi802293m
|
| [45] |
Devereux M, Gresh N, Piquemal JP, et al. (2014) A supervised fitting approach to force field parametrization with application to the SIBFA polarizable force field. J Comput Chem 35: 1577–1591. doi: 10.1002/jcc.23661
|
| [46] |
Wu R, Lu Z, Cao Z, et al. (2011) A transferable nonbonded pairwise force field to model zinc interactions in metalloproteins. J Chem Theory Comput 7: 433–443. doi: 10.1021/ct100525r
|
| [47] |
Sakharov DV, Lim C (2005) Zn protein simulations including charge transfer and local polarization effects. J Am Chem Soc 127: 4921–4929. doi: 10.1021/ja0429115
|
| [48] |
Chakravorty DK, Wang B, Lee CW, et al. (2012) Simulations of allosteric motions in the zinc sensor CzrA. J Am Chem Soc 134: 3367–3376. doi: 10.1021/ja208047b
|
| [49] |
Chakravorty DK, Parker TM, Guerra AJ, et al. (2013) Energetics of zinc-mediated interactions in the allosteric pathways of metal sensor proteins. J Am Chem Soc 135: 30–33. doi: 10.1021/ja309170g
|
| [50] |
Reyes-Caballero H, Campanello GC, Giedroc DP (2011) Metalloregulatory proteins: Metal selectivity and allosteric switching. Biophys Chem 156: 103–114. doi: 10.1016/j.bpc.2011.03.010
|
| [51] |
Andrews CT, Elcock AH (2013) Molecular dynamics simulations of highly crowded amino acid solutions: comparisons of eight different force field combinations with experiment and with each other. J Chem Theory Comput 9: 4585–4602. doi: 10.1021/ct400371h
|
| [52] |
Abriata LA, Dal Peraro M (2015) Assessing the potential of atomistic molecular dynamics simulations to probe reversible protein-protein recognition and binding. Sci Rep 5: 10549. doi: 10.1038/srep10549
|
 biophy-05-01-077-s1.pdf biophy-05-01-077-s1.pdf |
 |
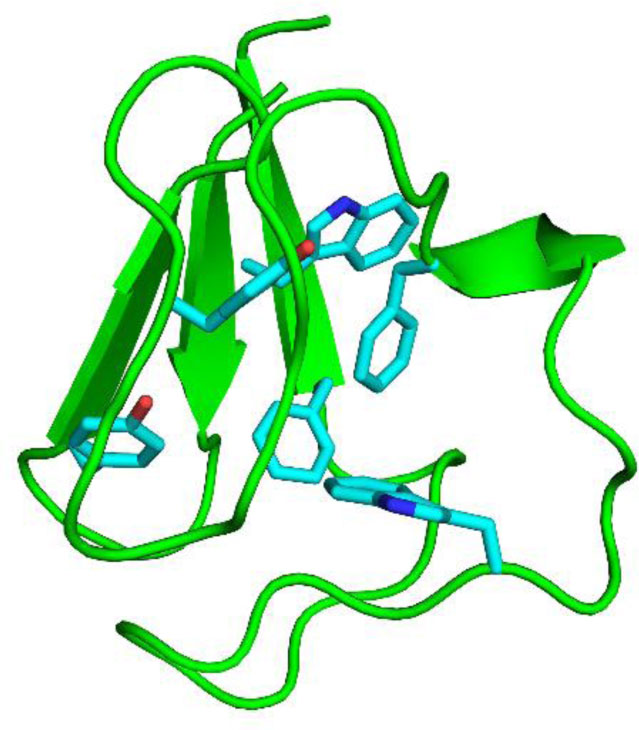
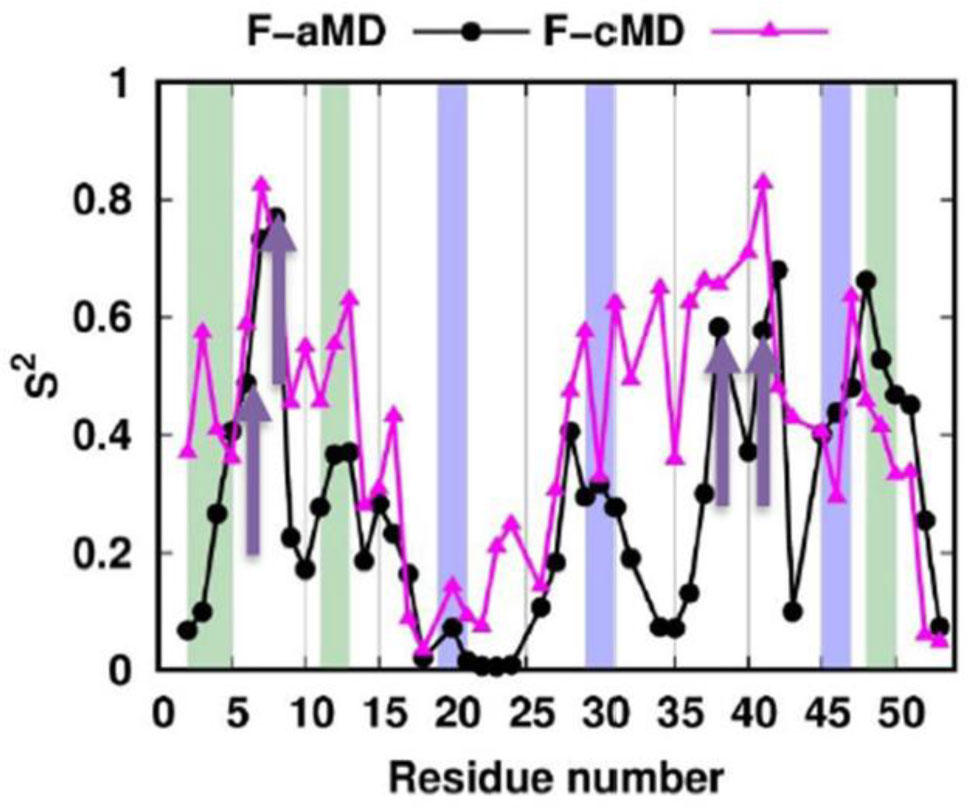
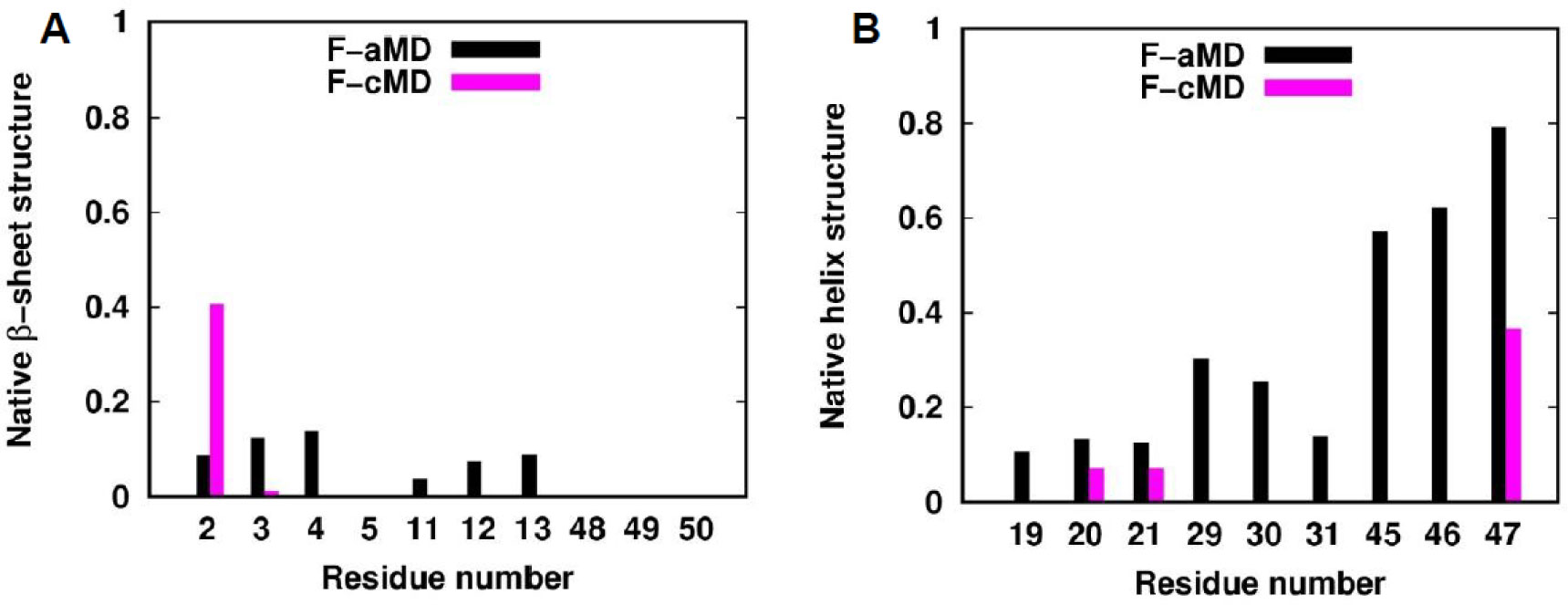
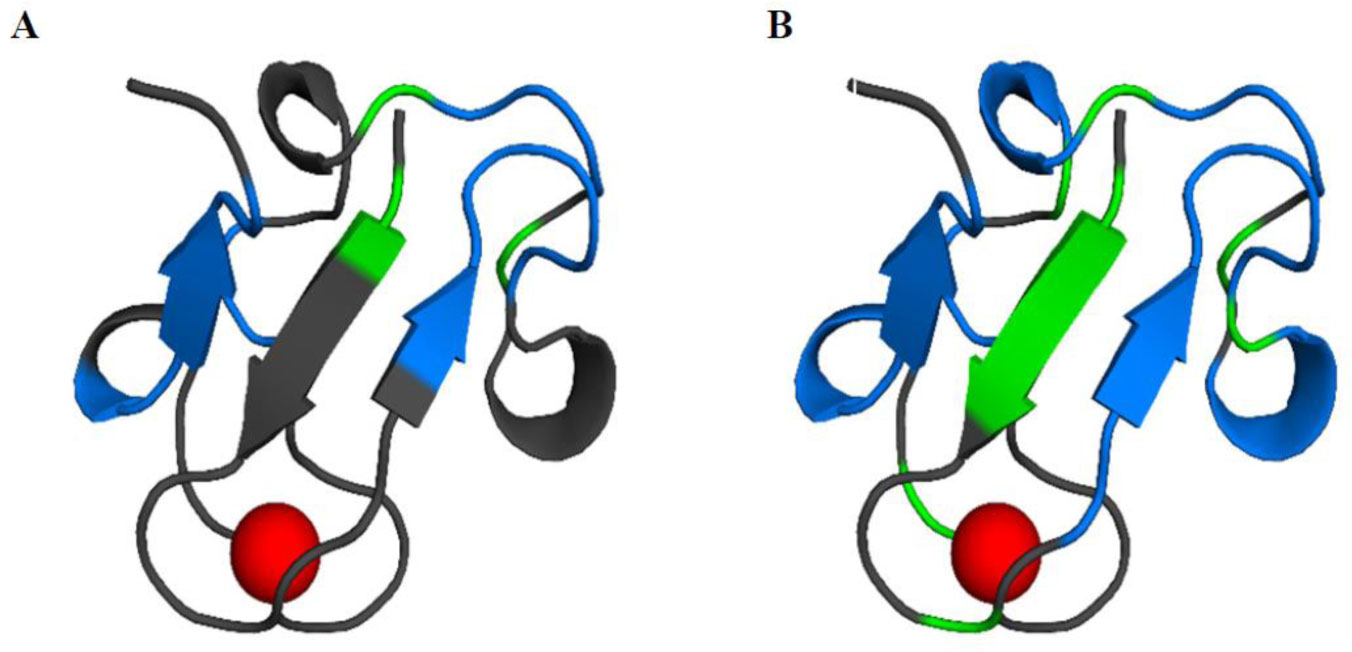
Figures(11) / Tables(2)

Davide Sala, Andrea Giachetti, Antonio Rosato. Molecular dynamics simulations of metalloproteins: A folding study of rubredoxin from Pyrococcus furiosus[J]. AIMS Biophysics, 2018, 5(1): 77-96. doi: 10.3934/biophy.2018.1.77







 DownLoad:
DownLoad: