Citation: Mohammad Azizur Rahman, Shahdat Hossain, Noorlidah Abdullah, Norhaniza Aminudin. Brain proteomics links oxidative stress with metabolic and cellular stress response proteins in behavioural alteration of Alzheimer’s disease model rats[J]. AIMS Neuroscience, 2019, 6(4): 299-315. doi: 10.3934/Neuroscience.2019.4.299

| [1] |
Reiman EM (2014) Alzheimer's disease and other dementias: advances in 2013. Lancet Neurol 13: 3–5. doi: 10.1016/S1474-4422(13)70257-6

|
| [2] |
Liao L, Cheng D, Wang J, et al. (2004) Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem 279: 37061–37068. doi: 10.1074/jbc.M403672200
|
| [3] |
Wang Q, Wu J, Rowan MJ, et al. (2005) β‐amyloid inhibition of long‐term potentiation is mediated via tumor necrosis factor. Eur J Neurosci 22: 2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x
|
| [4] | Lecanu L, Papadopoulos V (2013) Modeling Alzheimer's disease with non-transgenic rat models. Alzheimers Res Ther 5: 17. |
| [5] |
Sparks DL, Scheff SW, Hunsaker JC, et al. (1994) Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126: 88–94. doi: 10.1006/exnr.1994.1044
|
| [6] |
Refolo LM, Malester B, LaFrancois J, et al. (2000) Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: 321–331. doi: 10.1006/nbdi.2000.0304
|
| [7] |
Anstey KJ, Lipnicki DM, Low LF (2008) Cholesterol as a risk factor for dementia and cognitive decline: A systematic review of prospective studies with meta-analysis. Am J Geriatr Psychiatry 16: 343–354. doi: 10.1097/01.JGP.0000310778.20870.ae
|
| [8] |
Ansari MA, Scheff SW (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69: 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
|
| [9] |
McLellan ME, Kajdasz ST, Hyman BT, et al. (2003) In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J Neurosci 23: 2212–2217. doi: 10.1523/JNEUROSCI.23-06-02212.2003
|
| [10] |
Li F, Calingasan NY, Yu F, et al. (2004) Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem 89: 1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x
|
| [11] |
Dumont M, Wille E, Stack C, et al. (2009) Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J 23: 2459–2466. doi: 10.1096/fj.09-132928
|
| [12] |
Murakami K, Murata N, Noda Y, et al. (2011) SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem 286: 44557–44568. doi: 10.1074/jbc.M111.279208
|
| [13] |
Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95. doi: 10.1152/physrev.00018.2001
|
| [14] |
Tramutola A, Lanzillotta C, Perluigi M, et al. (2017) Butterfield, Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull 133: 88–99. doi: 10.1016/j.brainresbull.2016.06.005
|
| [15] | Mamun AA, Hashimoto M, Katakura M, et al. (2014) neuroprotective effect of madecassoside evaluated using amyloid Β 1-42 -mediated in vitro and in vivo Alzheimer's disease models. Intl J Indigenous Med Plants 47: 1669–1682. |
| [16] |
Olton DS, Samuelson RJ (1976) Remembrance of places passed: spatial memory in rats. J Exp Psychol Anim Behav Process 2: 97–116. doi: 10.1037/0097-7403.2.2.97
|
| [17] |
Jarrard LE, Okaichi H, Steward O, et al. (1984) On the role of hippocampal connections in the performance of place and cue tasks: comparisons with damage to hippocampus. Behav Neurosci 98: 946–954. doi: 10.1037/0735-7044.98.6.946
|
| [18] |
Shevchenko G, Sjödin MO, Malmström D, et al. (2010) Cloud-point extraction and delipidation of porcine brain proteins in combination with bottom-up mass spectrometry approaches for proteome analysis. J Proteome Res 9: 3903–3911. doi: 10.1021/pr100116k
|
| [19] | Stepanichev MY, Zdobnova IM, Zarubenko II, et al. (2007) Studies of the effects of central administration of β-amyloid peptide (25–35): pathomorphological changes in the hippocampus and impairment of spatial memory. Neurosci Behav Physiol 36: 101–106. |
| [20] |
Holscher C, Gengler S, Gault VA, et al. (2007) Soluble beta-amyloid[25-35] reversibly impairs hippocampal synaptic plasticity and spatial learning. Eur J Pharmacol 561: 85–90. doi: 10.1016/j.ejphar.2007.01.040
|
| [21] |
Parihar MS, Brewer GJ (2007) Mitoenergetic failure in Alzheimer disease. Am J Physiol Cell Physiol 292: C8–C23. doi: 10.1152/ajpcell.00232.2006
|
| [22] |
Butterfield DA, Hardas SS, Lange ML (2010) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer's disease: many pathways to neurodegeneration. J Alzheimers Dis 20: 369–393. doi: 10.3233/JAD-2010-1375
|
| [23] |
Minjarez B, Valero Rustarazo ML, Sanchez del Pino MM, et al. (2013) Identification of polypeptides in neurofibrillary tangles and total homogenates of brains with Alzheimer's disease by tandem mass spectrometry. J Alzheimers Dis 34: 239–262. doi: 10.3233/JAD-121480
|
| [24] |
Musunuri S, Wetterhall M, Ingelsson M, et al. (2014) Quantification of the brain proteome in Alzheimer's disease using multiplexed mass spectrometry. J Proteome Res 13: 2056–2068. doi: 10.1021/pr401202d
|
| [25] |
Butterfield DA, Perluigi M, Reed T, et al. (2012) Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal 17: 1610–1655. doi: 10.1089/ars.2011.4109
|
| [26] |
Poon HF, Calabrese V, Calvani M, et al. (2006) Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal 8: 381–394. doi: 10.1089/ars.2006.8.381
|
| [27] |
Díez‐Vives C, Gay M, García‐Matas S, et al. (2009) Proteomic study of neuron and astrocyte cultures from senescence‐accelerated mouse SAMP8 reveals degenerative changes. J Neurochem 111: 945–955. doi: 10.1111/j.1471-4159.2009.06374.x
|
| [28] |
Blair LJ, Zhang B, Dickey CA (2013) Potential synergy between tau aggregation inhibitors and tau chaperone modulators. Alzheimers Res Ther 5: 41–50. doi: 10.1186/alzrt207
|
| [29] |
Yao J, Irwin RW, Zhao L, et al. (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. PNAS 106: 14670–14675. doi: 10.1073/pnas.0903563106
|
| [30] |
Cui Y, Huang M, He Y, et al. (2011) Genetic ablation of apolipoprotein A-IV accelerates Alzheimer's disease pathogenesis in a mouse model. Am J Pathol 178: 1298–1308. doi: 10.1016/j.ajpath.2010.11.057
|
| [31] |
Keeney JTR, Swomley AM, Förster S, et al. (2013) Apolipoprotein A‐I: Insights from redox proteomics for its role in neurodegeneration. Proteomics Clin Appl 7: 109–122. doi: 10.1002/prca.201200087
|
| [32] |
Liu HC, Hu CJ, Chang JG, et al. (2006) Proteomic identification of lower apolipoprotein AI in Alzheimer's disease. Dement Geriatr Cogn Disord 21: 155–161. doi: 10.1159/000090676
|
| [33] |
Corder EH, Saunders AM, Strittmatter WJ, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923. doi: 10.1126/science.8346443
|
| [34] |
Sizova D, Charbaut E, Delalande F, et al. (2007) Proteomic analysis of brain tissue from an Alzheimer's disease mouse model by two-dimensional difference gel electrophoresis. Neurobiol Aging 28: 357–370. doi: 10.1016/j.neurobiolaging.2006.01.011
|
| [35] |
Ellis RJ, Olichney JM, Thal LJ, et al. (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: The CERAD experience, part XV. Neurology 46: 1592–1596. doi: 10.1212/WNL.46.6.1592
|
| [36] |
Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer's disease. Neuron 63: 287–303. doi: 10.1016/j.neuron.2009.06.026
|
| [37] | Choi J, Gao J, Kim J, et al. (2015) The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Aβ amyloidosis. Sci Transl Med 7: 314ra184. |
| [38] |
Tosto G, Reitz C (2013) Genome-wide association studies in Alzheimer's disease: A review. Curr Neurol Neurosci Rep 13: 381. doi: 10.1007/s11910-013-0381-0
|
| [39] |
Yu JT, Tan L (2012) The role of clusterin in Alzheimer's disease: pathways, pathogenesis and therapy. Mol Neurobiol 45: 314–326. doi: 10.1007/s12035-012-8237-1
|
| [40] |
Hong I, Kang T, Yoo Y, et al. (2013) Quantitative proteomic analysis of the hippocampus in the 5XFAD mouse model at early stages of Alzheimer's disease pathology. J Alzheimers Dis 36: 321–334. doi: 10.3233/JAD-130311
|
| [41] |
Puglielli L, Konopka G, Pack-Chung E, et al. (2001) Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat Cell Biol 3: 905–912. doi: 10.1038/ncb1001-905
|
| [42] |
Sjögren M, Mielke M, Gustafson D, et al. (2006) Cholesterol and Alzheimer's disease-is there a relation? Mech Ageing Dev 127: 138–147. doi: 10.1016/j.mad.2005.09.020
|
| [43] |
Cortes-Canteli M, Zamolodchikov D, Ahn HJ, et al. (2012) Fibrinogen and altered hemostasis in Alzheimer's disease. J Alzheimers Dis 32: 599–608. doi: 10.3233/JAD-2012-120820
|
| [44] |
Ahn HJ, Zamolodchikov D, Cortes-Canteli M, et al. (2010) Alzheimer's disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A 107: 21812–21817. doi: 10.1073/pnas.1010373107
|
| [45] |
Li X, Buxbaum JN (2011) Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer's disease? Mol Neurodegener 6: 79. doi: 10.1186/1750-1326-6-79
|
| [46] |
Flynn JM, Melov S (2013) SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic Biol Med 62: 4–12. doi: 10.1016/j.freeradbiomed.2013.05.027
|
| [47] |
De Leo ME, Borrello S, Passantino M, et al. (1998) Oxidative stress and overexpression of manganese superoxide dismutase in patients with Alzheimer's disease. Neurosci Lett 250: 173–176. doi: 10.1016/S0304-3940(98)00469-8
|
| [48] |
Manavalan A, Mishra M, Sze SK, et al. (2013) Brain-site-specific proteome changes induced by neuronal P60TRP expression. Neurosignals 21: 129–149. doi: 10.1159/000343672
|
| [49] |
Sun KH, Chang KH, Clawson S, et al. (2011) Glutathione‐S‐transferase P1 is a critical regulator of Cdk5 kinase activity. J Neurochem 118: 902–914. doi: 10.1111/j.1471-4159.2011.07343.x
|
| [50] |
Luo J, Wärmländer SKTS, Gräslund A, et al. (2014) Non-chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid β peptide. J Biol Chem 289: 27766–27775. doi: 10.1074/jbc.M114.574947
|
| [51] |
Habib KL, Lee TCM, Yang J (2010) Inhibitors of catalase-amyloid interactions protect cells from β-amyloid-induced oxidative stress and toxicity. J Biol Chem 285: 38933–38943. doi: 10.1074/jbc.M110.132860
|
| [52] |
Cocciolo A, Di Domenico F, Coccia R, et al. (2012) Decreased expression and increased oxidation of plasma haptoglobin in Alzheimer disease: Insights from redox proteomics. Free Radic Biol Med 53: 1868–1876. doi: 10.1016/j.freeradbiomed.2012.08.596
|
| [53] |
Spagnuolo MS, Maresca B, La Marca V, et al. (2014) Haptoglobin interacts with apolipoprotein E and beta-amyloid and influences their crosstalk. ACS Chem Neurosci 5: 837–847. doi: 10.1021/cn500099f
|
| [54] |
Poynton AR, Hampton MB (2014) Peroxiredoxins as biomarkers of oxidative stress. Biochim Biophys Acta 1840: 906–912. doi: 10.1016/j.bbagen.2013.08.001
|
 neurosci-06-04-299-s001.pdf neurosci-06-04-299-s001.pdf |
 |

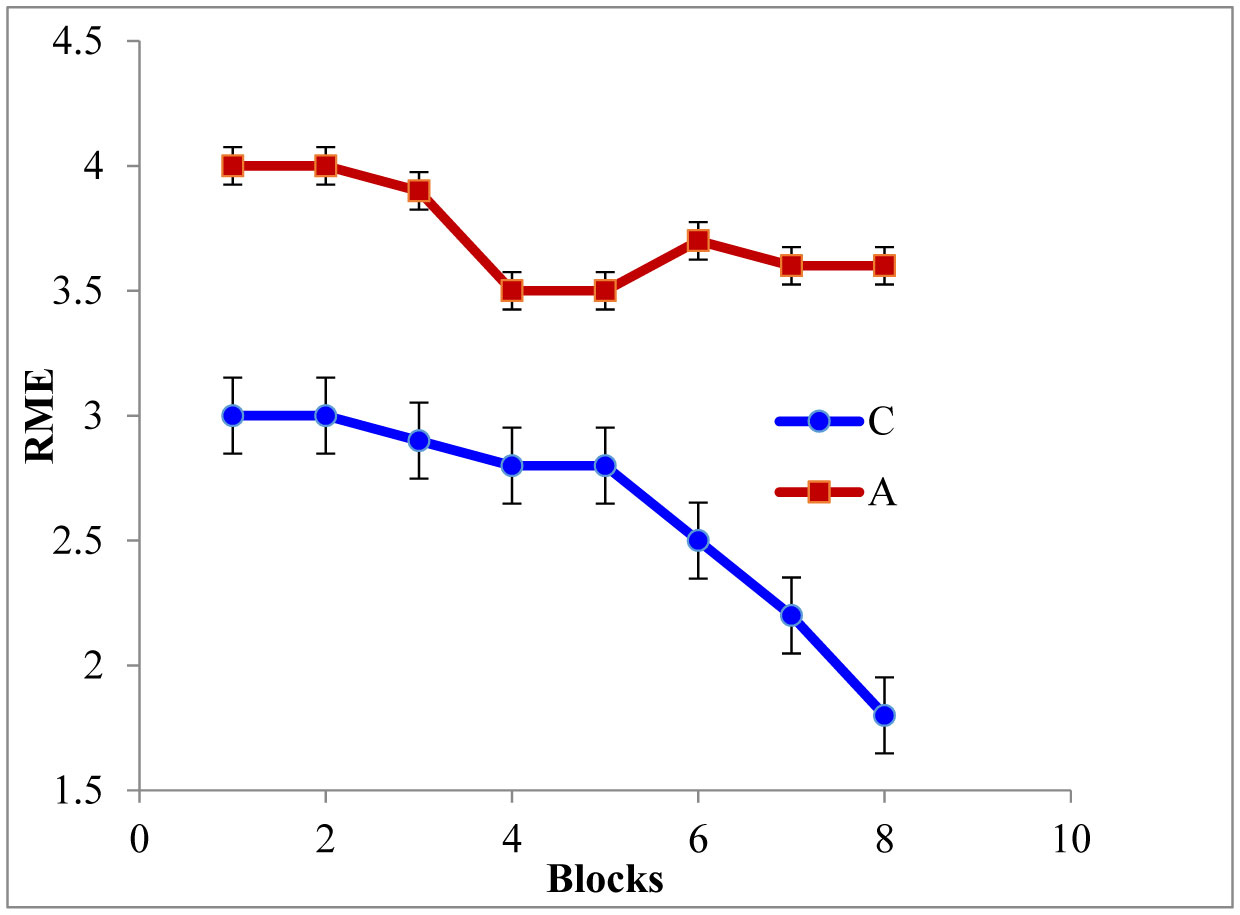
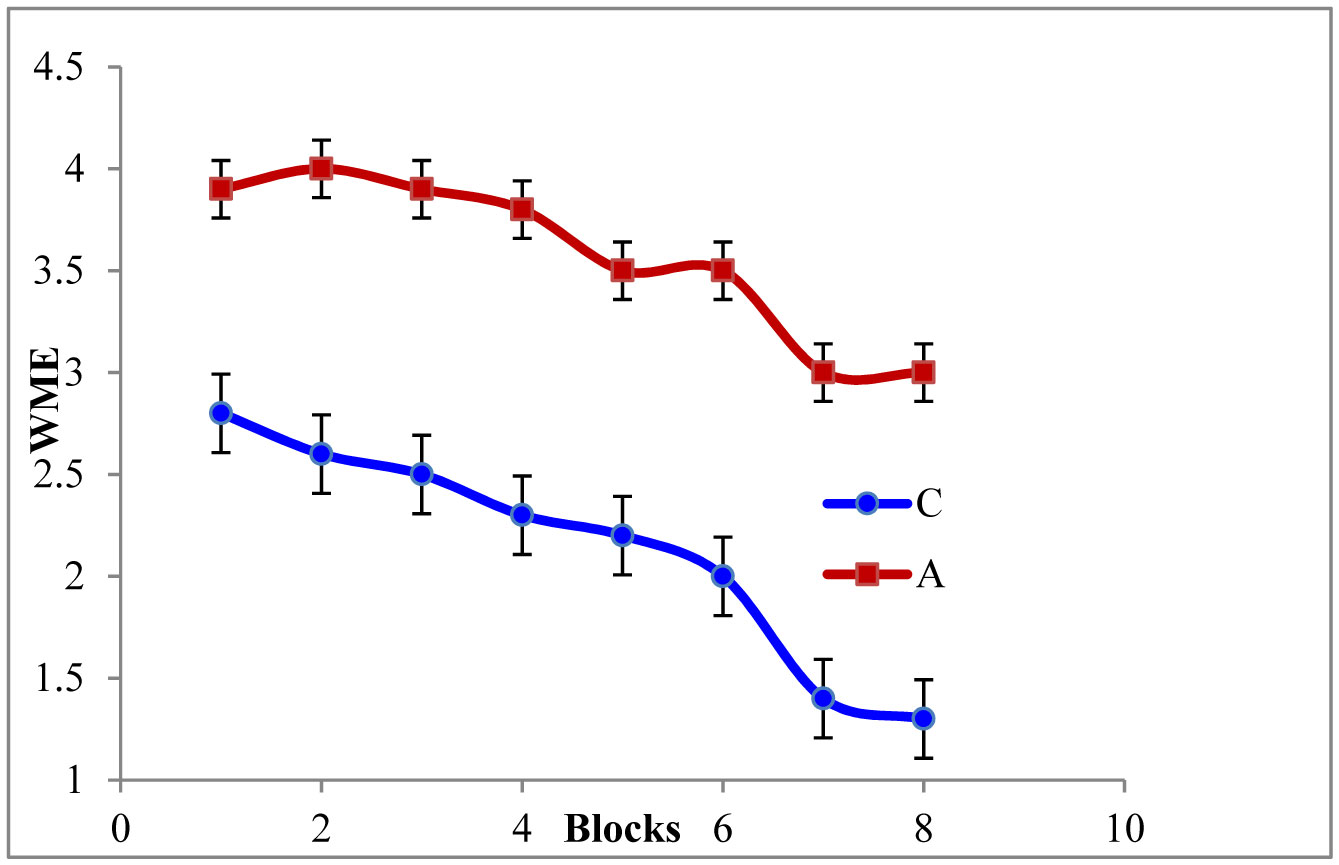
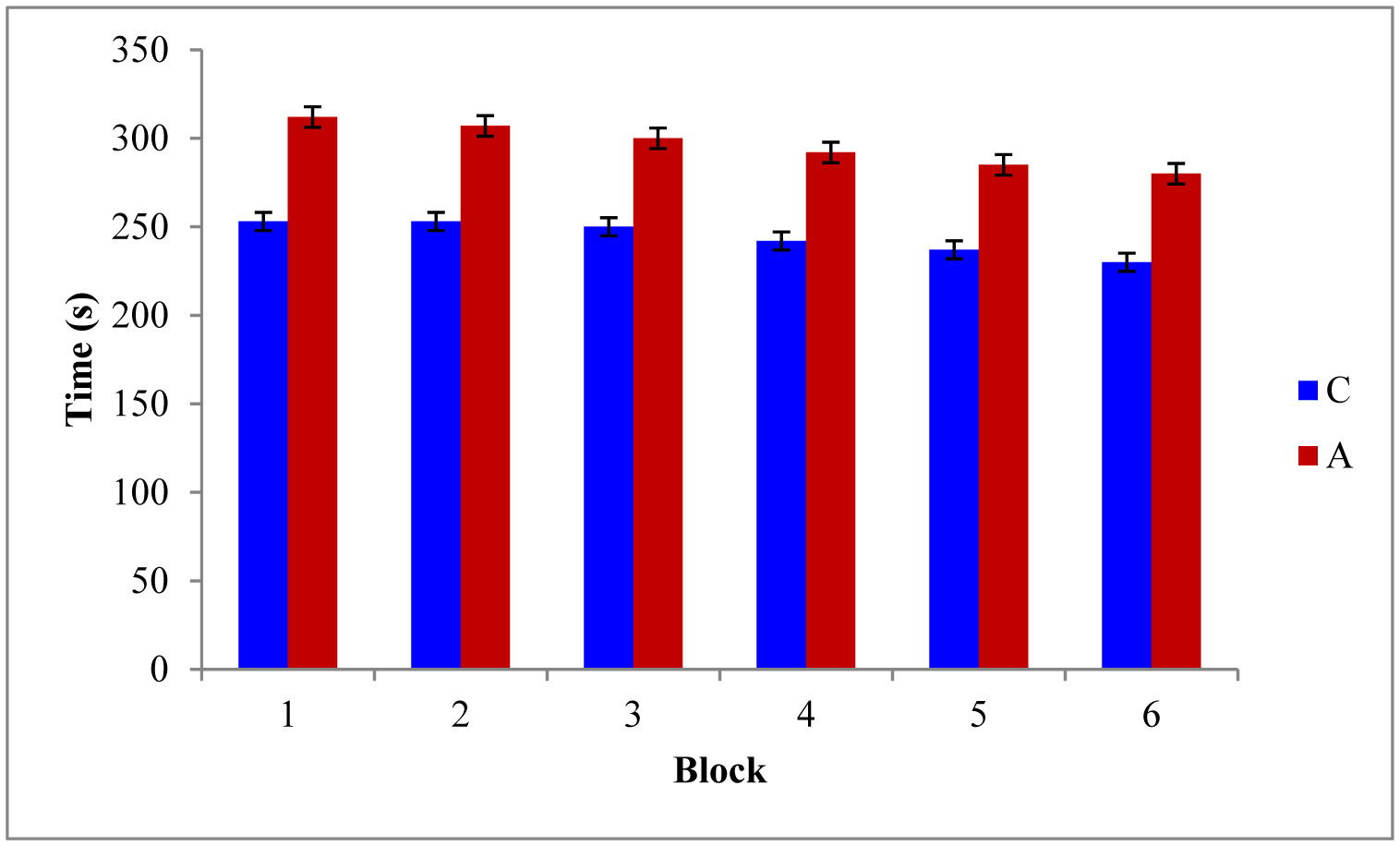
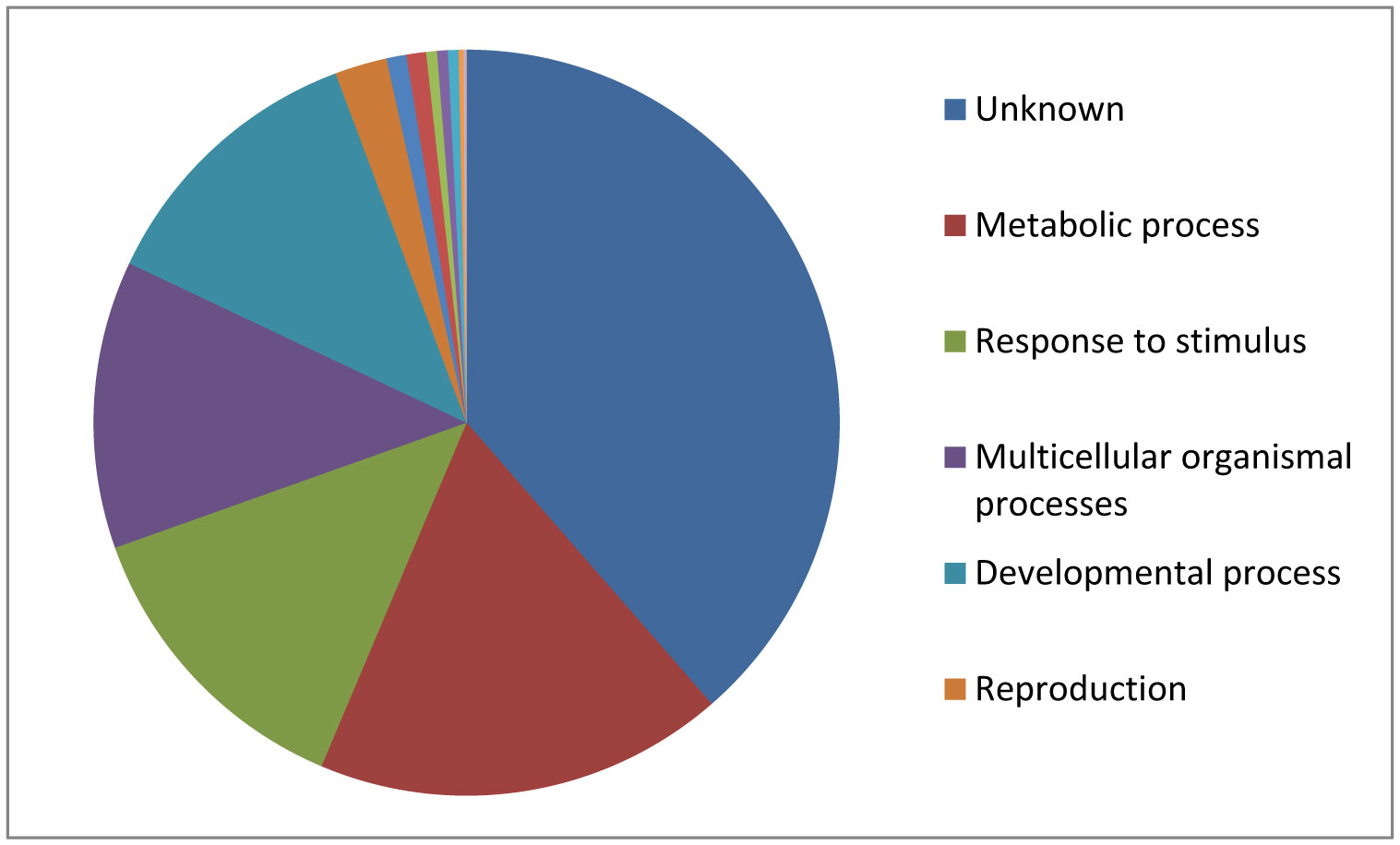
Figures(5)

Mohammad Azizur Rahman, Shahdat Hossain, Noorlidah Abdullah, Norhaniza Aminudin. Brain proteomics links oxidative stress with metabolic and cellular stress response proteins in behavioural alteration of Alzheimer’s disease model rats[J]. AIMS Neuroscience, 2019, 6(4): 299-315. doi: 10.3934/Neuroscience.2019.4.299







 DownLoad:
DownLoad: