Citation: Zenab Arooj Sher, Karen J Liu. Congenital tracheal defects: embryonic development and animal models[J]. AIMS Genetics, 2016, 3(1): 60-73. doi: 10.3934/genet.2016.1.60

| [1] |
Cohen MM Jr., Kreiborg S (1992) Upper and lower airway compromise in the Apert syndrome. Am J Med Genet 44: 90-93. doi: 10.1002/ajmg.1320440121

|
| [2] | Kim JH, Kim PCW, Hui C-c (2001) The VACTERL association: lessons from the Sonic hedgehog pathway. Clin Genet 59: 306-315. |
| [3] |
Elleru RG, Whitsett JA (2004) Potential role of Sox9 in patterning tracheal cartilage ring formation in an embryonic mouse model. Arch Otolaryngol Head Neck Surg 130: 732- 736. doi: 10.1001/archotol.130.6.732
|
| [4] |
Macchiarini P, Jungebluth P, Go T, et al. (2008) Clinical transplantation of a tissue-engineered airway. The Lancet 372: 2023-2030. doi: 10.1016/S0140-6736(08)61598-6
|
| [5] | Kaufman MH (1992) The Atlas of Mouse Development: Academic Press Inc. 512. |
| [6] |
Perl AK, Wert SE, Nagy A, et al. (2002) Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A 99: 10482-10487. doi: 10.1073/pnas.152238499
|
| [7] |
Park J, Zhang JJ, Moro A, et al. (2010) Regulation of Sox9 by Sonic Hedgehog (Shh) is essential for patterning and formation of tracheal cartilage. Dev Dyn 239: 514-526. doi: 10.1002/dvdy.22192
|
| [8] |
Que J, Choi M, Ziel JW, et al. (2006) Morphogenesis of the trachea and esophagus: current players and new roles for noggin and Bmps. Differentiation 74: 422-437. doi: 10.1111/j.1432-0436.2006.00096.x
|
| [9] | Lim FY, Crombleholme TM, Hedrick HL, et al. (2003) Congenital high airway obstruction syndrome: natural history and management. J Pediatr Surg 38: 940-945. |
| [10] |
Hirose S, Harrison MR (2003) The ex utero intrapartum treatment (EXIT) procedure. Seminars in Neonatology 8: 207-214. doi: 10.1016/S1084-2756(03)00029-0
|
| [11] | Hirakawa H, Ueno S, Yokoyama S, et al. (2002) Tracheal agenesis: a case report. Tokai J Exp Clin Med 27: 1-7. |
| [12] |
Masters IB (2009) Congenital airway lesions and lung disease. Pediatr Clin North Am 56: 227-242. doi: 10.1016/j.pcl.2008.10.006
|
| [13] | Austin J, Ali T (2003) Tracheomalacia and bronchomalacia in children: pathophysiology, assessment, treatment and anaesthesia management. Paediatric Anaesthesia 13: 3-11. |
| [14] |
Burden RJ (1999) Tracheobronchial malacia and stenosis in children in intensive care: bronchograms help to predict outcome. Thorax 54: 511-517. doi: 10.1136/thx.54.6.511
|
| [15] |
Herrera P, Caldarone C, Forte V, et al. (2007) The current state of congenital tracheal stenosis. Pediatr Surg Int 23: 1033-1044. doi: 10.1007/s00383-007-1945-3
|
| [16] | Noorily MR, Farmer DL, Belenky WM, et al. (1999) Congenital Tracheal Anomalies in the Craniosynostosis Syndromes. J Paediatr Surg 34: 1036-1039. |
| [17] |
Eswarakumar VP, Horowitz MC, Locklin R, et al. (2004) A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A 101: 12555-12560. doi: 10.1073/pnas.0405031101
|
| [18] |
Lertsburapa K, Schroeder JW Jr., Sullivan C (2010) Tracheal cartilaginous sleeve in patients with craniosynostosis syndromes: a meta-analysis. J Pediatr Surg 45: 1438-1444. doi: 10.1016/j.jpedsurg.2009.09.005
|
| [19] |
Ioannides AS, Massa V, Ferraro E, et al. (2010) Foregut separation and tracheo-oesophageal malformations: the role of tracheal outgrowth, dorso-ventral patterning and programmed cell death. Dev Biol 337: 351-362. doi: 10.1016/j.ydbio.2009.11.005
|
| [20] |
Merei JM, Hutson JM (2002) Embryogenesis of tracheo esophageal anomalies: a review. Pediatr Surg Int 18: 319-326. doi: 10.1007/s00383-002-0751-1
|
| [21] | Mahlapuu M, Enerback S, Carlsson P (2001) Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128: 2397-2406. |
| [22] |
Arora R, Metzger RJ, Papaioannou VE (2012) Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet 8: e1002866. doi: 10.1371/journal.pgen.1002866
|
| [23] | Minoo P, Guoshan S, Drum H, et al. (1999) Defects in tracheoesophageal and lung morphogenesis in Nkx2.1 (−/−) mouse embryos. Developmental Biology 209: 60-71. |
| [24] | Mendelsohn C, Lohnes D, Decimo D, et al. (1994) Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120: 2749-2771. |
| [25] |
Domyan ET, Ferretti E, Throckmorton K, et al. (2011) Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 138: 971-981. doi: 10.1242/dev.053694
|
| [26] |
Miller LA, Wert SE, Clark JC, et al. (2004) Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev Dyn 231: 57-71. doi: 10.1002/dvdy.20105
|
| [27] |
Motoyama J, Liu J, Mo R, et al. (1998) Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nature Genetics 20: 54-57. doi: 10.1038/1711
|
| [28] |
Thomas HM, Todd PJ, Heaf D, et al. (1994) Recurrence of Pallister-Hall syndrome in two sibs. J Med Genet 31: 145-147. doi: 10.1136/jmg.31.2.145
|
| [29] |
Zakin L, Metzinger CA, Chang EY, et al. (2008) Development of the vertebral morphogenetic field in the mouse: interactions between Crossveinless-2 and Twisted Gastrulation. Dev Biol 323: 6-18. doi: 10.1016/j.ydbio.2008.08.019
|
| [30] |
Sala FG, Del Moral PM, Tiozzo C, et al. (2011) FGF10 controls the patterning of the tracheal cartilage rings via Shh. Development 138: 273-282. doi: 10.1242/dev.051680
|
| [31] |
Rock JR, Futtner CR, Harfe BD (2008) The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol 321: 141-149. doi: 10.1016/j.ydbio.2008.06.009
|
| [32] |
Suemoto H, Muragaki Y, Nishioka K, et al. (2007) Trps1 regulates proliferation and apoptosis of chondrocytes through Stat3 signaling. Dev Biol 312: 572-581. doi: 10.1016/j.ydbio.2007.10.001
|
| [33] |
Tiozzo C, De Langhe S, Carraro G, et al. (2009) Fibroblast Growth Factor 10 plays a causative role in the tracheal cartilage defects in a mouse model of Apert Syndrom. Paediatric Research 66: 386-390. doi: 10.1203/PDR.0b013e3181b45580
|
| [34] |
Elluru RG, Thompson F, Reece A (2009) Fibroblast growth factor 18 gives growth and directional cues to airway cartilage. Laryngoscope 119: 1153-1165. doi: 10.1002/lary.20157
|
| [35] |
Regnier CH, Masson R, Kedinger V, et al. (2002) Impaired neural tube closure, axial skeleton malformations, and tracheal ring disruption in TRAF4-deficient mice. Proc Natl Acad Sci USA 99: 5585-5590. doi: 10.1073/pnas.052124799
|
| [36] |
Hines EA, Jones MK, Verheyden JM, et al. (2013) Establishment of smooth muscle and cartilage juxtaposition in the developing mouse upper airways. Proc Natl Acad Sci USA 110: 19444-19449. doi: 10.1073/pnas.1313223110
|
| [37] |
Pole RJ, Qi BQ, Beasley SW (2001) Abnormalities of the tracheal cartilage in the rat fetus with tracheo-oesophageal fistula or trachea agenesis. Peadiatric Surg Int 17: 25-28. doi: 10.1007/s003830000448
|
| [38] | Gonfiotti A, Jaus MO, Barale D, et al. (2013) The first tissue-engineered airway transplantation: 5-year follow-up results. The Lancet 383: 238-244. |
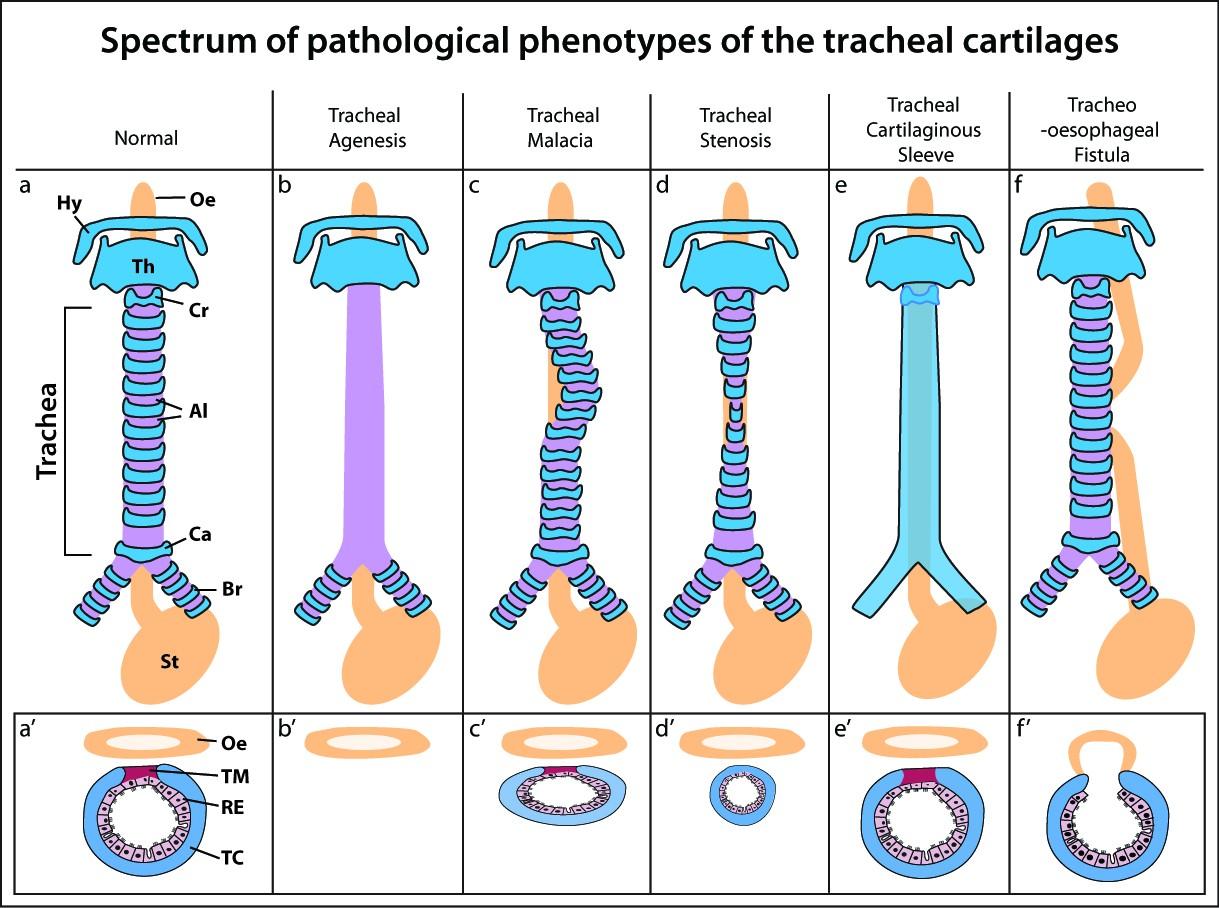
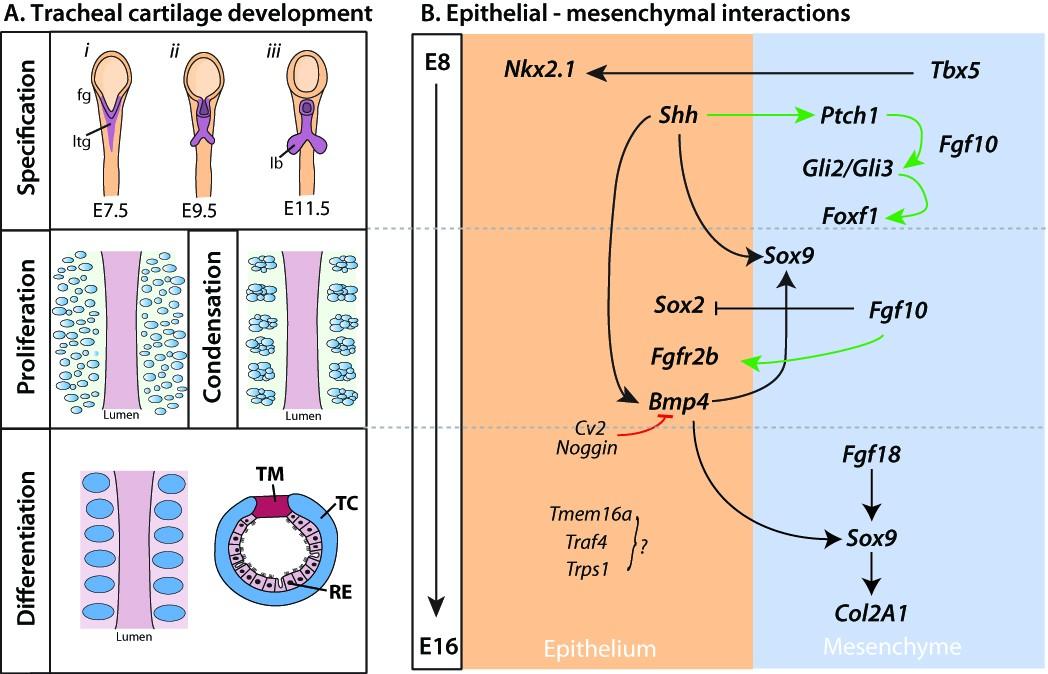
Figures(2) / Tables(1)

Zenab Arooj Sher, Karen J Liu. Congenital tracheal defects: embryonic development and animal models[J]. AIMS Genetics, 2016, 3(1): 60-73. doi: 10.3934/genet.2016.1.60







 DownLoad:
DownLoad: